

























-
砷来源广泛,包括火山喷发、岩石风化等自然来源以及采矿、冶金等人为来源[1-2]。在全球范围内,土壤中砷的平均质量分数为1.8 mg·kg−1,而我国土壤中砷平均质量分数达到9.2 mg·kg−1,超过世界水平的5倍[3]。我国云南、贵州、四川等西南地区的土壤中砷背景值远超全国土壤背景值[4]。土壤中的砷通过食物链进入人体后,可引发色素沉着、慢性肺病、心血管疾病和神经系统紊乱等健康问题[5]。因此,对砷污染土壤的修复十分迫切。
电动修复是常用的一种砷污染土壤修复方法,其利用电渗析、电迁移等电动效应使砷酸根和亚砷酸根定向迁移,从而降低土壤中砷的总量[6-7]。但常规电动修复技术对砷的修复效果有限,KARACA等[8]对沉积物中的砷进行电动修复时发现,运行18 d后砷几乎没有被去除。电极逼近法为电动修复的一种,其在电动过程中每隔一段时间将电极向某一方向移动一定距离,以此来影响土壤pH、氧化还原电位 (Eh) 等环境因子,而砷的溶解性和迁移性与环境因子密切相关。YAO等[9]发现,相比于固定电极法 (FE-EK) ,阴极逼近法 (AC-EK) 通过提高阴极区域pH可将砷的去除率提高4倍。付博等[10]发现,当pH<4时,随着pH的降低,粗砂和细砂中砷的溶出量不断增加。周一敏等[11]发现,当Eh较低时,五价砷[As(V)]会转化为移动性更高的三价砷[As(III)],另外还能驱动土壤中砷的释放。由此可见,电动逼近技术对提高砷污染土壤修复效果具有很大潜力。
目前,常采用向土壤中加入化学药剂[2,7]、增设渗透反应墙[6,12]等方式提高砷去除率,但基于电极逼近技术对砷污染土壤进行修复的研究尚很缺乏。基于此,本研究采用不同的电极逼近方式对砷污染土壤进行修复。研究不同逼近方式对总砷[As(T)]的分布以及As(III)、As(V)迁移转化的影响,探究捕集室土壤中砷赋存形态的转化,以期为砷污染场地修复提供技术参考和理论依据。
-
供试土壤采自辽宁大连某污染场地,经风干并研磨后过20目标准筛备用。供试土壤基本理化性质为:pH为7.17,Eh为273.5 mV,电导率为2 012.5 μS·cm−1,Al、Fe、Ca质量分数分别为40.13、44.17、102.55 g·kg−1,As(T)、As(III)、As(V)质量分数分别为355.08、120.32、234.76 mg·kg−1。其中,As(T)质量分数超过《土壤环境质量 建设用地土壤污染风险管控标准 (试行) 》 (GB 36600-2018) [13]第一类用地筛选值 (20 mg·kg−1) 的17倍。
-
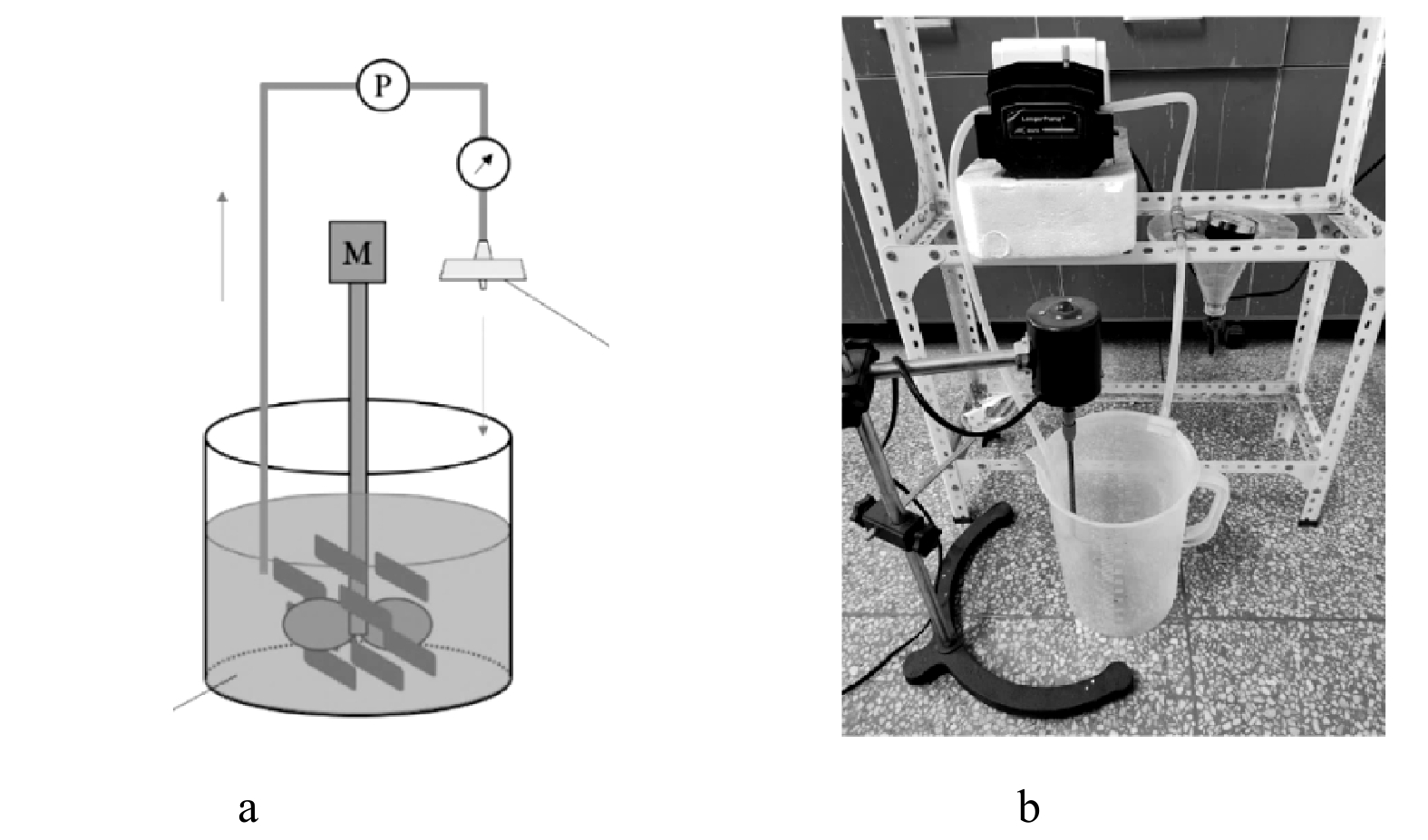
如图1(a)所示,实验装置主体由土壤室和捕集室组成,捕集室置于实验装置中部,可自由取出,2侧为土壤室。取样点位置如图1(b)所示,从阳极到阴极划分为阳极区 (S1~S3) 、捕集室 (S4) 、阴极区 (S5~S7) 3部分,S1~S7每个区域设置3个取样点,将3个取样点的土壤混合后作为该区域的代表性土壤。
-
实验共设置4个电动处理组,分别为FE-EK、AC-EK、阳极逼近处理组 (AA-EK) 、两极逼近处理组 (AAC-EK) 。其中,FE-EK处理组不移动电极;AC-EK处理组的阴极电极每隔10 d向阳极方向移动4 cm,共移动2次;AA-EK处理组的阳极电极每隔10 d向阴极方向移动4 cm,共移动2次;AAC-EK处理组的阳极电极和阴极电极每隔10 d相向各移动4 cm,共移动2次。各电动处理组土壤室内均填装1 600 g污染土壤,捕集室内填装400 g混有质量分数为20% Fe2O3的污染土壤。另取400 g混有Fe2O3的污染土壤,不通电,作为电动处理组的对照组 (CK) 。
实验以不锈钢电极为电极,电压恒定为24 V,处理时间30 d。实验过程中每隔4~5 d采用重量法补充去离子水,保持土壤含水率为30%。取样间隔为10 d,移动电极后的无电场区域不再继续取样。
-
本研究中总能耗和单位修复能耗的计算方法见式(1)和式(2)[14]。
式中:E为总能耗,kWh;U为实验电压,V;I为电流,A;t为修复时间,h。
式中:E0为单位修复能耗,kWh·mg−1;c0和c30为第0 天和第30天时捕集室中As(T)质量分数,mg·kg−1;m为捕集室中土壤质量,kg。
-
电流使用电流监控装置监测并记录。pH和电导率使用pH计 (PHS-3C型,上海仪电科学仪器股份有限公司) 和电导率仪 (CON700,美国Eutech公司) 测定[6]。Eh参考《土壤 氧化还原电位的测定 电位法》 (HJ 746—2015) [15],使用便携式ORP测定仪 (TR-901型,上海仪电科学仪器股份有限公司) 测定。As(T)质量分数利用HNO3-HF-HClO4对土壤进行分步消解[16],并用电感耦合等离子体质谱仪 (ICAPRQ,美国Thermo Fisher Scientific公司) 测定。As(III)质量分数参考ZHENG等[17]以及张静等[18]的提取方法,并用原子荧光光谱仪 (AFS-9700A,北京海光仪器有限公司) 测定[19]。As(V)质量分数为As(T)与As(III)的差值。砷赋存形态参考XU等[7]的方法依次提取可交换态砷 (Ex-As) 、铝结合态砷 (Al-As) 、铁结合态砷 (Fe-As) 以及钙结合态砷 (Ca-As) ,并用ICP-OES (Avio 220 Max,美国PerkinElmer公司) 测定。残渣态砷 (Res-As) 测定方法同As(T)。
-
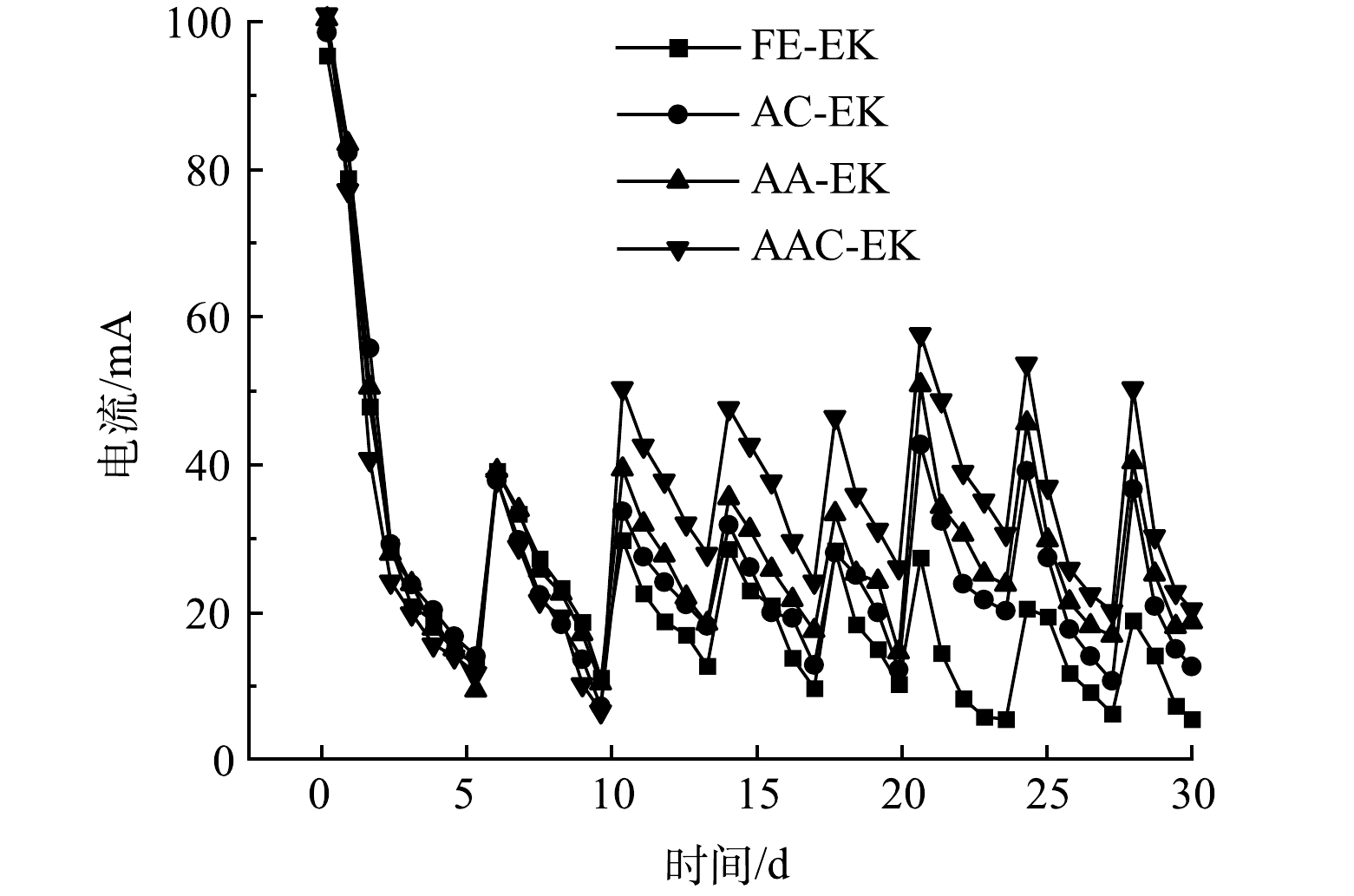
如图2所示,各处理组在移动电极前的电流值相似,表明各处理组间的平行性较好。通电后电流在短时间内即达到最大值,约为100 mA;随后电流值迅速下降,至第5 d时仅为9.42~14.04 mA;第5 d补水后电流值又迅速上升。这是因为,电动初始时土壤中含有大量可移动离子;而后电解水产生的H+和OH−被不断中和,孔隙水中的离子强度降低[9];补水后土壤中的可移动离子数量又有所增加 [20-21]。电导率常用来表示土壤孔隙液中溶解离子的数量[22]。各处理组的电导率变化如图3所示,表现为两极高、中间低。这归因于阴离子和阳离子不断迁往两极[6],降低了中间区域可溶性离子数量。各处理组电导率在S1、S4、S5区域存在显著差异 (p<0.05) 。
运行10 d后,电极逼近处理组的电流值高于固定电极处理组,以第20 d为例,FE-EK、AC-EK、AA-EK以及AAC-EK的电流值分别为27.36、42.64、50.74、57.68 mA。这主要是因为,随着电极的移动,土壤有效长度缩短,提高了系统电流[9]。因AAC-EK的两极间距最短,所以AAC-EK的电流值又高于AC-EK和AA-EK。AC-EK的电流值低于AA-EK主要是因为AC-EK能提高阴极区pH,容易生成氢氧化物、碳酸盐等不溶性和非导电物质,降低系统电流[21]。
-
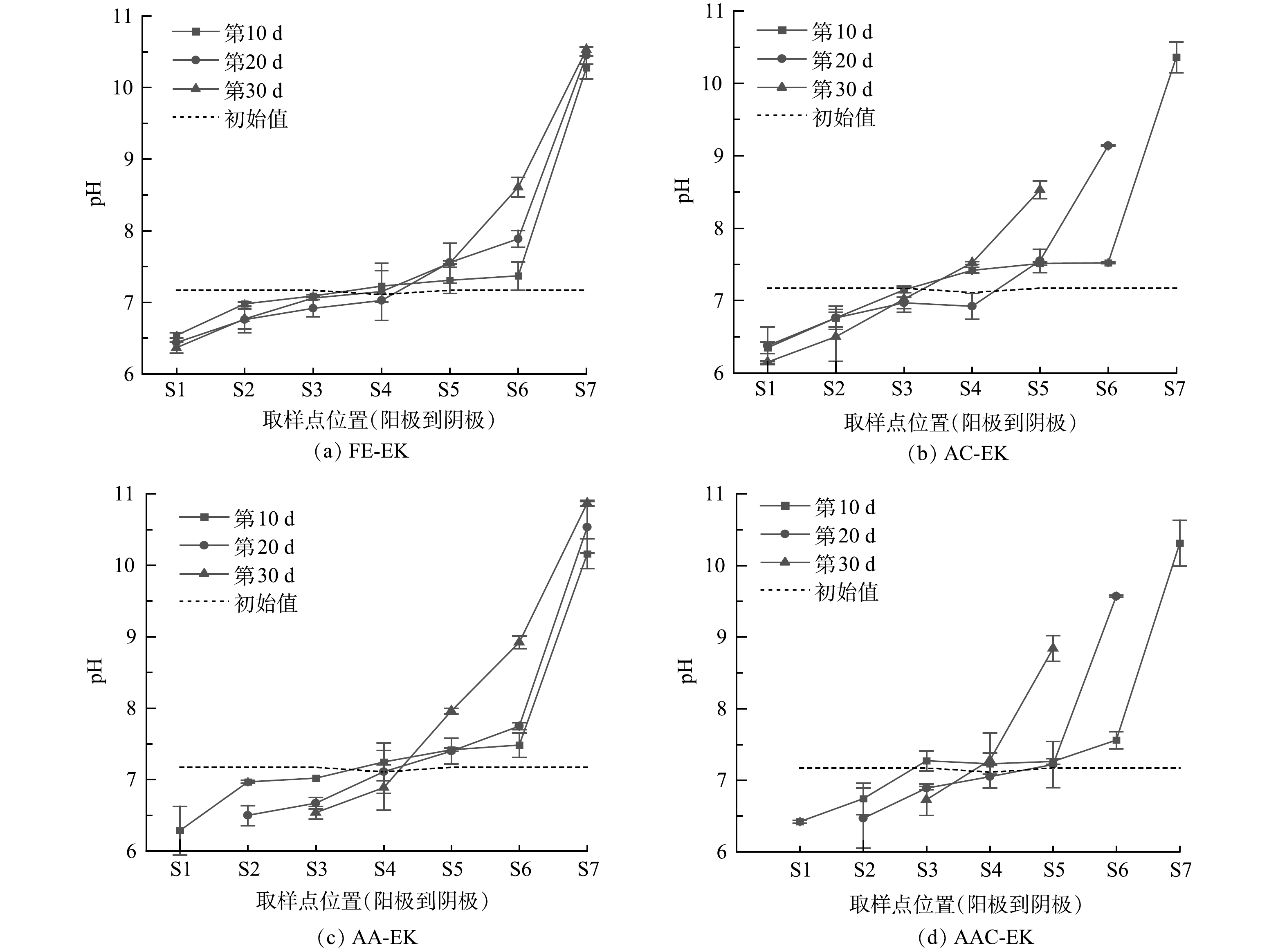
土壤室土壤初始pH为7.17,捕集室土壤初始pH为7.11。如图4所示,土壤pH从阳极至阴极呈逐渐增大趋势,且阴极区变化幅度高于阳极区。这是因为,在外加电场作用下,阳极和阴极因发生水解反应分别生成H+和OH−[9]。AA-EK能够促进阳极区pH降低,例如其S2区域在10~20 d降低0.47,高于FE-EK下降幅度,但AA-EK并未能阻止阴极区的pH升高,这可能是由于土壤的酸缓冲性能较高,向阴极移动的H+在到达阴极区前就被消耗殆尽。反之,AC-EK的阴极电极不断向阳极靠近,使其阴极区pH随时间的推移逐渐升高。由于AAC-EK电流值最高,导致其S2~S6区域的pH变化幅度一般高于AC-EK、AA-EK或FE-EK。
-
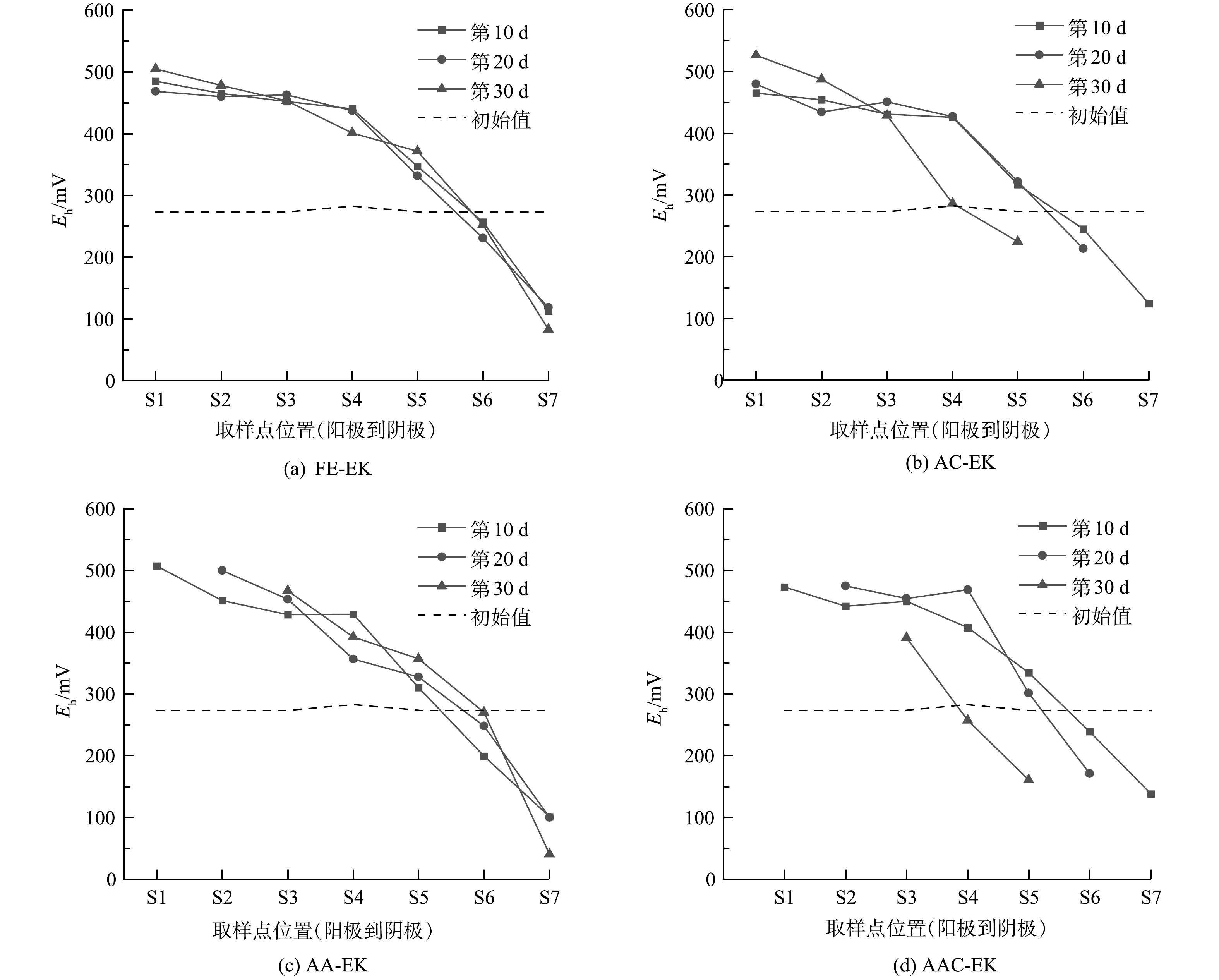
如图5所示,土壤室土壤初始Eh为273.5 mV,捕集室土壤初始Eh为282.5 mV。电动结束后,土壤Eh表现为从阳极到阴极逐渐降低的分布趋势。其中,S1~S5区域的Eh一般高于初始值,S6~S7区域的Eh低于初始值。阳极Eh的升高主要源于水电解反应产生的氧气及活性自由基;而阴极Eh的降低主要源于水解反应产生氢气,使阴极土壤处于还原气氛。与FE-EK相比,阳极电极的移动促进阳极区Eh升高,而阴极电极的移动促进阴极区Eh降低。以AC-EK为例,其第30 d时S5区域的Eh比FE-EK低147 mV,与SHEN等[23]的研究结果一致。
-
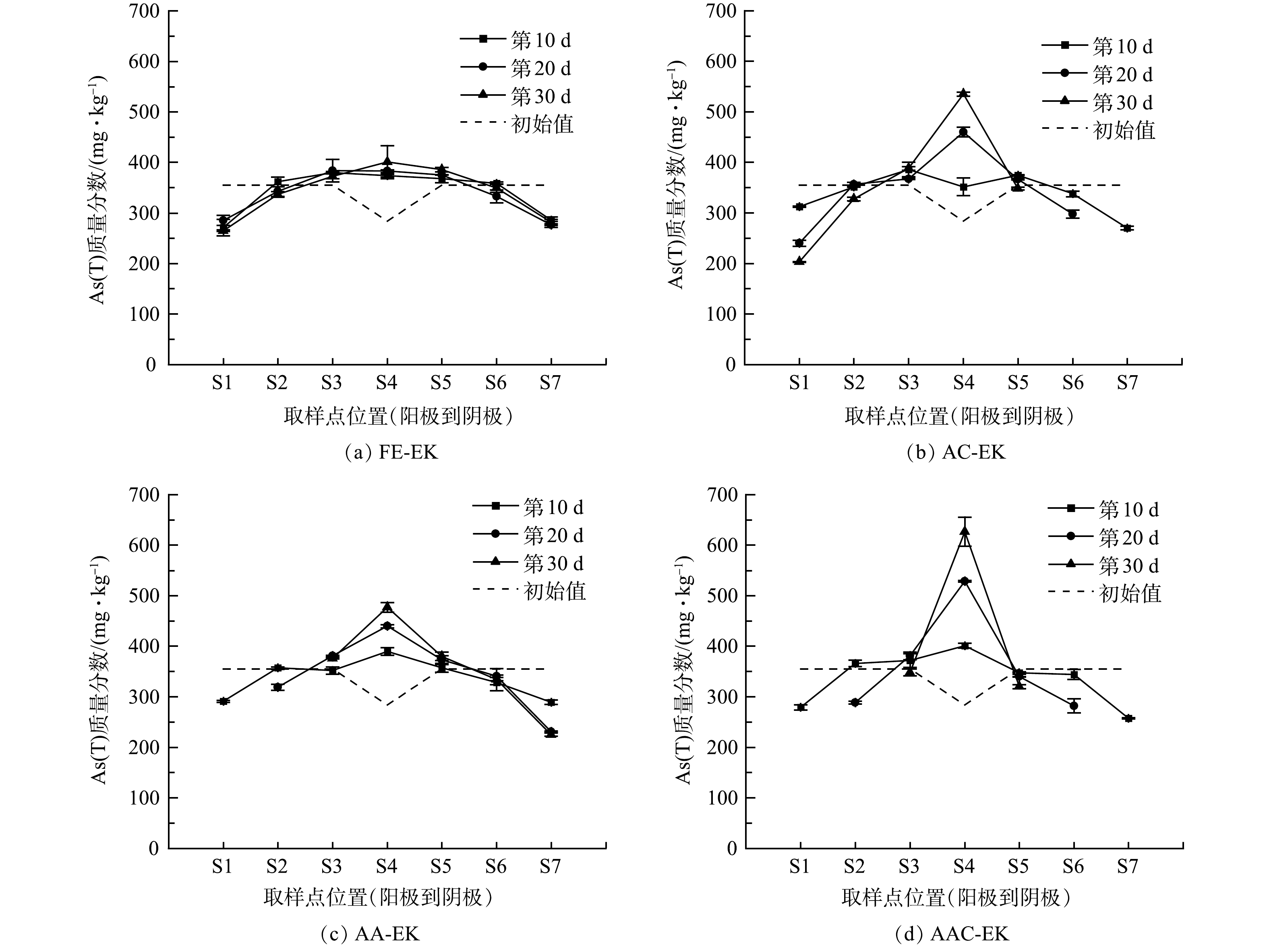
如图6所示,土壤室土壤初始As(T)质量分数为355.08 mg·kg−1,捕集室土壤初始As(T)质量分数为283.97 mg·kg−1。修复过程中,土壤中As(T)从两极区域向中间区域聚集,并最终呈现两极低、中间高的分布趋势。As(T)分布的变化是因为,As(T)在电场作用下同时受到电迁移和电渗析作用,一方面,带负电荷的H2AsO4−、HAsO42−、H2AsO3−等随电迁移迁往阳极;另一方面,溶解于土壤孔隙水中的砷随电渗流迁往阴极[24],导致两极及其附近区域As(T)质量分数降低。由于土壤中对砷吸附能力较强的铝、铁、钙等元素较多,可与砷形成不可移动的沉淀,导致砷移动性显著降低;此外,捕集室中Fe2O3对砷具有很强的吸附能力,迁移至此的砷难以继续向两极迁移,使得捕集室中As(T)质量分数不断升高。运行30 d后,AC-EK、AA-EK以及AAC-EK捕集室中As(T)质量分数与初始值相比显著升高 (p<0.05) ,S1、S7区域As(T)质量分数显著性降低 (p<0.05),以AAC-EK处理组As(T)质量分数显著性降低点位最多 (S1、S2、S5、S6、S7) ,而FE-EK捕集室中As(T)质量分数与初始值相比无显著性差异 (p>0.05) ,仅S1区域As(T)显著性降低 (p<0.05) ,这表明电极逼近对As(T)的迁移具有显著促进作用。
运行30 d后,FE-EK的As(T)整体迁移率最低 (15.38%) ,AAC-EK的As(T)整体迁移率最高 (31.50%) ,AC-EK与AA-EK居于2者之间 (27.25%、21.65%) 。AC-EK之所以能促进砷的迁移主要因为以下几个方面:首先,电极间距的缩短增大了系统电流,加速了砷的迁移;其次,阴极电极的移动增大了阴极区土壤pH,使土壤对带负电荷的砷酸根和亚砷酸根吸附能力减弱[2],且OH−能置换出以含氧阴离子形式存在的砷[25];此外,阴极电极的移动还降低了土壤Eh,使Fe(III)向Fe(II)转化,Fe(OH)3等铁系物因此发生溶解[26],砷因失去吸附相被释放到土壤溶液中,有利于砷的迁移。AA-EK因电极间距的缩短增大了系统电流,同样能促进As(T)的迁移;但因为其阳极区域pH不断降低,增强了土壤对砷的吸附,导致促进效果不明显。虽然AAC-EK阳极区pH也较低,但它的电流值最高,且其阴极区pH最高,Eh最低,有利于砷的解吸,所以AAC-EK对As(T)的迁移效果优于AC-EK和AA-EK。
如图7所示,FE-EK、AC-EK、AA-EK、AAC-EK的总电能耗依次为373.46、449.59、496.46、572.64 kWh,单位修复能耗依次为7.98、4.47、6.44、4.18 kWh·mg−1。可见,总电能耗最高的AAC-EK的单位修复能耗最低。这是因为,当电压一定时,单位修复能耗除了与电流强度有关还与污染物迁移量有关,AAC-EK捕集室中的As(T)的增加量为FE-EK的2.93倍。
-
初始土壤中,As(V)为无机砷的主要形式,约为As(III)的1.95倍。电动处理30 d后As(V)的分布如图8(a)所示。As(V)表现为中间高、两极低的分布趋势,FE-EK、AC-EK、AA-EK、AAC-EK捕集室中As(V)质量分数依次升高60.62%、120.61%、93.99%、162.86%。这是因为,阴极带负电荷的As(V)不断移向阳极,在迁移过程中,pH逐渐降低,As(V)迁移能力随之下降;且中间区域的Fe2O3对As(V)有强亲和力,导致As(V)移动至捕集室后难以继续移动,并最终停滞在捕集室;另外,由于电渗析流会带动部分溶解于土壤间隙液中As(V)向阴极迁移,导致阳极区的As(V)也有不同程度的降低。各处理组间As(V)分布差异主要集中在阴极区,AC-EK和AAC-EK能够升高阴极区pH,进而提高砷的移动性,所以这2个处理组阴极区的As(V)质量分数低于AA-EK和FE-EK;又因为AA-EK电流较大,所以其阴极区的As(V)质量分数又低于FE-EK。
As(III)的分布如图8(b)所示。As(III)与As(V)分布趋势一致,为中间高、两极低。这是因为,阳极区土壤pH<9.2,As(III)以不带电的分子形式 (H3AsO3) 存在,主要受电渗析作用迁往阴极[27];在阴极区,越接近阴极土壤pH越高,As(III)又以分子形式向含氧酸根形式转化,带负电荷的亚砷酸根 (H2AsO3−、HAsO32−、AsO33−) 逐渐增多,并随电迁移迁往阳极,导致S6、S7区域的As(III)质量分数低于S5区域。对比来看,各处理组阳极区As(III)质量分数从低到高依次为AAC-EK、AA-EK、AC-EK、FE-EK。处理组间的差异可能与电流强度有关,当电流较大时电渗析作用较强,更多的As(III)受电渗析作用迁移向阴极,所以电流越大阳极区的As(III)残留量越低,同时使得捕集室中As(III)质量分数越高。
由于土壤Eh普遍升高,导致部分As(III)转化为As(V)。运行30 d后,FE-EK、AC-EK、AA-EK、AAC-EK各点位As(III)平均质量分数较初始值分别降低9.78%、7.81%、13.65%、4.09%。与此同时,As(V)质量分数随之升高。有研究指出,As(III)的毒性高于As(V)[3],因此,经电动修复土壤中砷的毒性被降低。比较而言,AA-EK因能提高阳极区Eh,所以对As(III)的削减量最高;AAC-EK虽然也能提高阳极区Eh,但其阴极区Eh明显降低,所以对As(III)的总体削减效果较差。
-
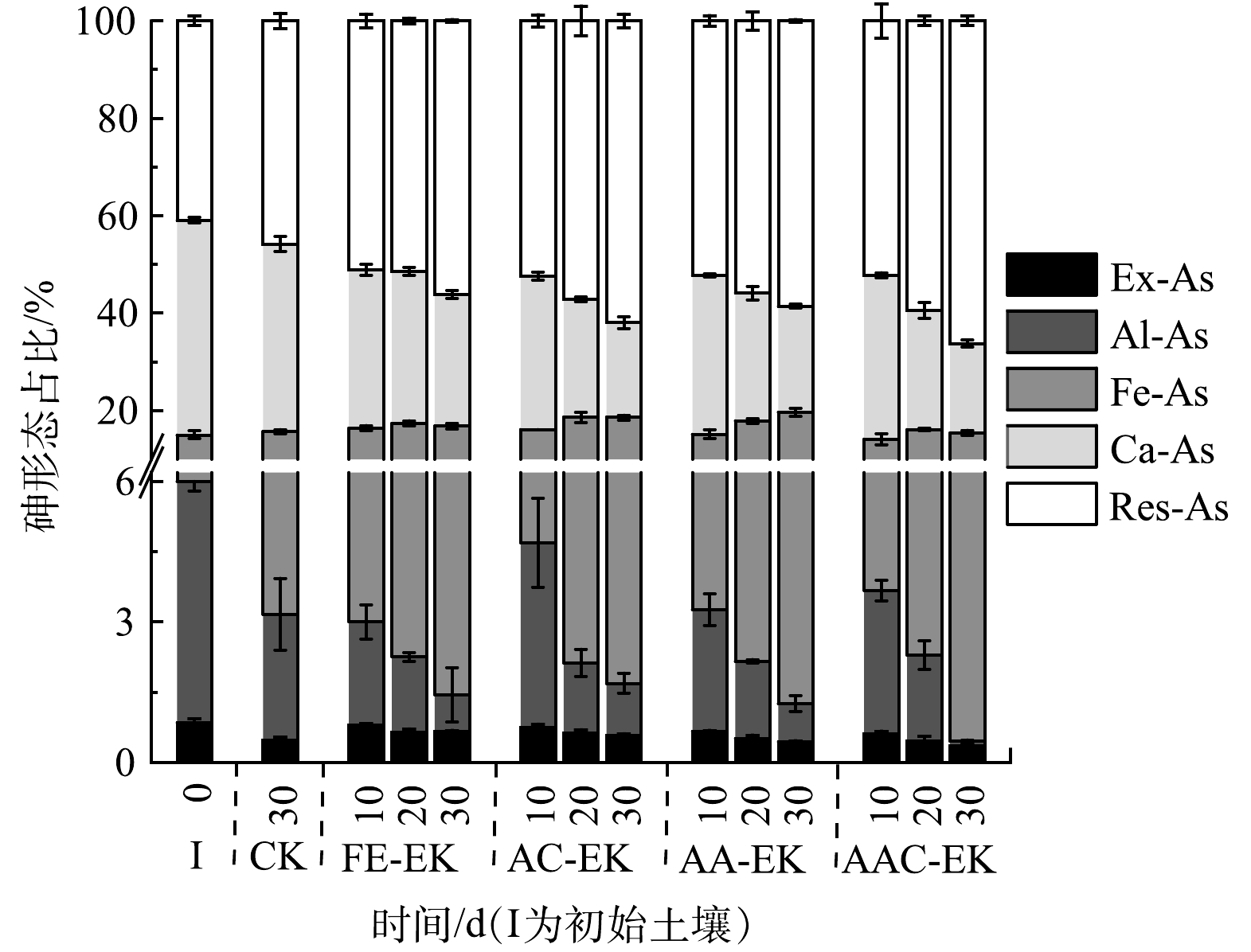
各处理组捕集室中砷的形态分布如图9所示。初始土壤中各形态砷占比从低到高依次为Ex-As (0.84%)、Al-As (5.16%)、Fe-As (9.05%)、Res-As (40.88%)、Ca-As (44.07%)。砷在电场的作用下不断向捕集室中迁移,并在Fe2O3的作用下发生赋存形态的明显转化,表现为Ex-As、Al-As、Ca-As占比下降,Fe-As和Res-As占比上升。对比各处理组砷赋存形态占比可知,FE-EK处理组的Ex-As最终占比最高,AA-EK、AAC-EK处理组的Ex-As最终占比较低,分别为0.44%和0.36%;FE-EK处理组的Res-As最终占比最低,AAC-EK处理组Res-As占比最终最高,达64.98%,为CK的1.42倍。
Ex-As占比的降低是因为Fe2O3的加入为砷提供了更多的吸附位点,使Ex-As转化为Fe-As。由于AA-EK、AAC-EK的电流较大,且阳极区pH相对较低,电极腐蚀后产生的Fe2+/Fe3+在随电迁移迁往阴极的过程中因pH逐渐增大而被沉淀于捕集室中,进一步加强了对Ex-As的吸附,导致其Ex-As占比较低。Al-As占比的降低也可能是受Fe2O3的影响。胡丽琼等[28]研究发现,当向砷污染水稻土中加入Fe2O3的量达到0.5 mg·kg−1时,Al-As已降至检测限以下。Res-As占比的升高一方面是由于Al-As、Ca-As向Res-As转化;另一方面,砷被铁吸附后形成Fe-As双核或单基配体化合物,或通过发生化学反应使沉淀于铁氧化物表面的砷酸盐形成砷酸铁沉淀,进而生成Res-As [29-31]。不同赋存形态砷的生物有效性从大到小依次为Ex-As>Ca-As>Al-As>Fe-As>Res-As[29]。可见,经电动修复后,砷的生物有效性大幅度降低。因AAC-EK处理组Ex-As占比最低,Res-As占比最高,所以处理效果最好。
-
1) 相比于固定电极,3种电极逼近方式通过影响环境因子 (pH、Eh) 以及系统电流,对As(T)的迁移具有促进作用,以AAC-EK的As(T)整体迁移率最高 (31.50%) ,且单位修复能耗最低。
2) 砷的价态转化受Eh影响,电动修复后,各处理组As(III)平均质量分数较初始值有所降低,As(V)平均质量分数较初始值有所升高。
3) 电动联合Fe2O3施用可使砷的形态从Ex-As、Al-As、Ca-As向Fe-As、Res-As转化,降低捕集室中的砷的生物有效性,以AAC-EK的稳定化效果最佳。由此可见,AAC-EK在修复砷污染土壤方面具备很大潜力。
电动修复过程中电极逼近对土壤砷迁移与形态转化的影响
Effect of the approaching electrode on the soil arsenic migration and speciation transformation during electrokinetic remediation
-
摘要: 以高浓度砷污染土壤为修复对象,探究电极逼近法耦合捕集室对砷污染土壤的修复效果。实验设置4个处理,分别为固定电极 (FE-EK) 、阴极逼近 (AC-EK) 、阳极逼近 (AA-EK) 和两极逼近 (AAC-EK) 。结果表明,AC-EK、AA-EK以及AAC-EK对总砷[As(T)]的迁移具有促进作用,表现为捕集室中As(T)质量分数与初始值相比显著升高 (p<0.05) ,而FE-EK捕集室中As(T)质量分数与初始值相比无显著性差异 (p>0.05) ,As(T)整体迁移率以AAC-EK最高 (31.50%) ,FE-EK最低 (15.38%) 。由于土壤整体氧化还原电位升高,使得FE-EK、AC-EK、AA-EK、AAC-EK处理组三价砷平均质量分数较初始值分别降低9.78%、7.81%、13.65%、4.09%。砷的生物有效性在Fe2O3和电动效应的联合作用下不断降低,表现为可交换态砷、铝结合态砷、钙结合态砷向铁结合态砷、残渣态砷转化。本研究结果表明,AAC-EK促进As(T)迁移的效果最好,可交换态砷占比最低,且单位修复能耗最低,具有良好的砷污染土壤修复潜力。Abstract: The remediation effect of the approaching electrode technique coupled with a capture chamber was evaluated using soil contaminated with a high concentration of arsenic. Four treatments were set up in the experiment: fixed-electrode electrokinetic (FE-EK), approaching cathode electrokinetic (AC-EK), approaching anode electrokinetic (AA-EK), and approaching anode and cathode electrokinetic (AAC-EK) techniques. The results demonstrated that AC-EK, AA-EK, and AAC-EK techniques promoted the migration of total arsenic [As(T)]: the mass fraction of As(T) in their capture chambers was significantly higher than the initial value(p<0.05), while that in the capture chamber of FE-EK did not differ significantly from the initial value(p>0.05). The overall migration rates of As(T) in AAC-EK was the highest (31.50%), and that in FE-EK was the lowest (15.38%). The average mass fraction of trivalent arsenic in FE-EK, AC-EK, AA-EK, and AAC-EK techniques decreased by 9.78%, 7.81%, 13.65%, and 4.09%, respectively, compared with the initial value owing to an increase in the overall soil redox potential. Under the combined influence of Fe2O3 and electric-influence, the bioavailability of arsenic was continuously reduced, as evidenced by the conversion of exchangeable arsenic, aluminum-bound arsenic, and calcium-bound arsenic to iron-bound arsenic and residual arsenic. This study showed that AAC-EK was associated with the highest As(T) migration, lowest proportion of exchangeable arsenic, and lowest energy consumption per unit of remediation. Thus, the AAC-EK technique had good potential for remediating arsenic-contaminated soil.
-
氨氮一般指水中以游离氨(
NH3 NH+4 废水脱氨主要方法有生物法、化学沉淀法、吹脱法、吸附法[5]。但是生物法流程长,反应器大,占地多,常需要额外投加碳源,能耗大,成本高;化学沉淀法成本高,再生难,有二次污染;吹脱法能耗大,有二次污染,出水氨氮浓度仍然偏高。吸附法具有除污效率高、操作便捷、适用范围广、吸附剂可重复使用等优点,在氨氮废水处理中逐渐得到了广泛关注。ALSHAMERI等[6]对比了高岭石、埃洛石、蒙脱石、海泡石等六种天然粘土矿物对
NH+4 NH+4 目前,大部分吸附材料多以微纳米颗粒的形式存在,直接与废水混合能够充分利用其吸附性能,但也存在微纳米材料使用中流失和回收困难等问题。通过微滤或超滤膜辅助回收,将会增加额外能耗和成本,影响其进一步的推广应用。也有研究将吸附材料制备成毫米级微球,通过柱吸附实现污染物去除,但大颗粒吸附材料吸附容量低于微纳米颗粒[7]。因此,如何既能保证吸附容量,又能实现微尺度吸附材料的低碳回收,已经成为吸附法应用中的新困难。动态膜技术具有成本低、操作压力小、清洗简单、微颗粒分离效果好等优点,为微尺度颗粒材料回收提供了绿色低碳新方法[8]。但目前多数研究围绕在动态膜系统过滤特性和应用效能,沸石吸附脱氨效能和优化改性,鲜有对动态膜成膜材料吸附性能的拓展和深度耦合探索,及对动态膜系统和成膜材料作用的独立解析,考虑到沸石吸附脱氨和动态膜分离的优点和不足,本研究尝试构建吸附与动态膜耦合系统,同步实现废水中氨氮去除和颗粒吸附材料回收,从而为吸附法进一步走向工程应用提供新方案。
本研究围绕吸附脱氨和动态膜回收2个方面,通过静态吸附实验,系统探究沸石吸附脱氨效能和投加量、初始氨氮浓度对脱氨效能的影响,揭示沸石吸附氨氮的动力学特性,阐明吸附-动态膜系统的脱氨效果和成膜特征,为沸石吸附脱氨和沸石颗粒回收的进一步应用提供参考。
1. 材料与方法
1.1 材料和实验用水
主要使用的实验材料包括:74~150 μm沸石,硅藻土,0.45 μm微孔滤膜,38 μm尼龙网,压力计等。实验所需试剂主要有氯化铵、酒石酸钾钠、纳氏试剂,均为分析纯。
在容量瓶中按照标准配置1 000 mg·L−1的氨氮储备溶液,配置完成后将溶液移入细口试剂瓶待用;使用时,按需求量取一定体积的氨氮储备溶液,按比例稀释为10 mg·L−1氨氮模拟废水。氨氮测定方法参考《纳氏试剂分光光度法》HJ 535-2009。
1.2 实验装置
为了保证同步进行氨氮去除和吸附材料的回收,使用自制循环吸附-动态膜系统进行成膜研究。循环吸附-动态膜系统和实验装置见图1,该装置由反应池、蠕动泵、自制膜组件、压力计、电动搅拌器组成。实验过程中,反应池中加入2 L的模拟废水及吸附剂,搅拌加速混合,通过蠕动泵将混合液送入自制膜组件,在膜组件中截留吸附剂并形成动态膜,膜分离后的混合液回流至反应池中。
1.3 动力学实验
称取0.4 g沸石移至150 mL锥形瓶中,取1 mL氨储备溶液于100 mL容量瓶中定容,配制模拟氨氮污水,初始质量浓度为10 mg·L−1,再分别转入锥形瓶;将锥形瓶同时放入恒温摇床中振荡(25 ℃,180 r min−1),反应进行至5、10、30 min和1、3、6、12、24 h时取出并取样7 mL左右于10 mL离心管中待滤;经0.45 μm滤膜过滤后测量滤液中氨氮浓度,进行准一级与准二级动力学拟合。
1.4 吸附等温实验
分别称取0.4 g沸石移至锥形瓶中,分别取1、2、4、6、8、10 mL氨储备溶液于100 mL容量瓶中定容,配制10、20、40、60、80、100 mg·L−1的模拟氨氮废水,按顺序移入锥形瓶中;将锥形瓶同时放入恒温摇床中振荡(25 ℃,180 r min−1),反应进行至5、10、30 min和1、3、6、12、24 h时取出并取样7 mL;经0.45 μm滤膜过滤后测氨氮浓度,计算去除率与吸附量,进行Langmuir与Freundlich拟合。
2. 结果与讨论
2.1 沸石吸附脱氨性能及吸附特性
1)沸石投加量对氨氮去除的影响。沸石投加量对氨氮的去除率及吸附量的影响结果如图2所示。可见,在100 mL体系中、25 ℃、180 r min−1恒温振荡箱和初始氨氮质量浓度10 mg·L−1反应条件下,沸石对模拟废水中氨氮的去除率随其投加量的增加逐渐增加,在投加量为10 g·L−1时氨氮去除率可到达67%左右,而当沸石投加量从1 g·L−1增加到10 g·L−1时,吸附量由1.48 mg·g−1降至0.64 mg·g−1。这是因为随着沸石投加量的增加,沸石可以提供更多的吸附位点和更大的比表面积,因此,氨氮的去除率随之增加,但吸附剂投加量的增加也会造成部分吸附位点被覆盖,吸附位点之间产生竞争,故单位质量的沸石的吸附效率随着吸附剂投加量的增大而逐渐降低。在低氨氮质量浓度为10 mg·L−1下开展的投加量优化实验中,沸石对氨氮去除效果较好。这是因为沸石是一种骨架状铝硅酸盐,具有离子交换的特性,对于
NH+4 NH+4 NH+4 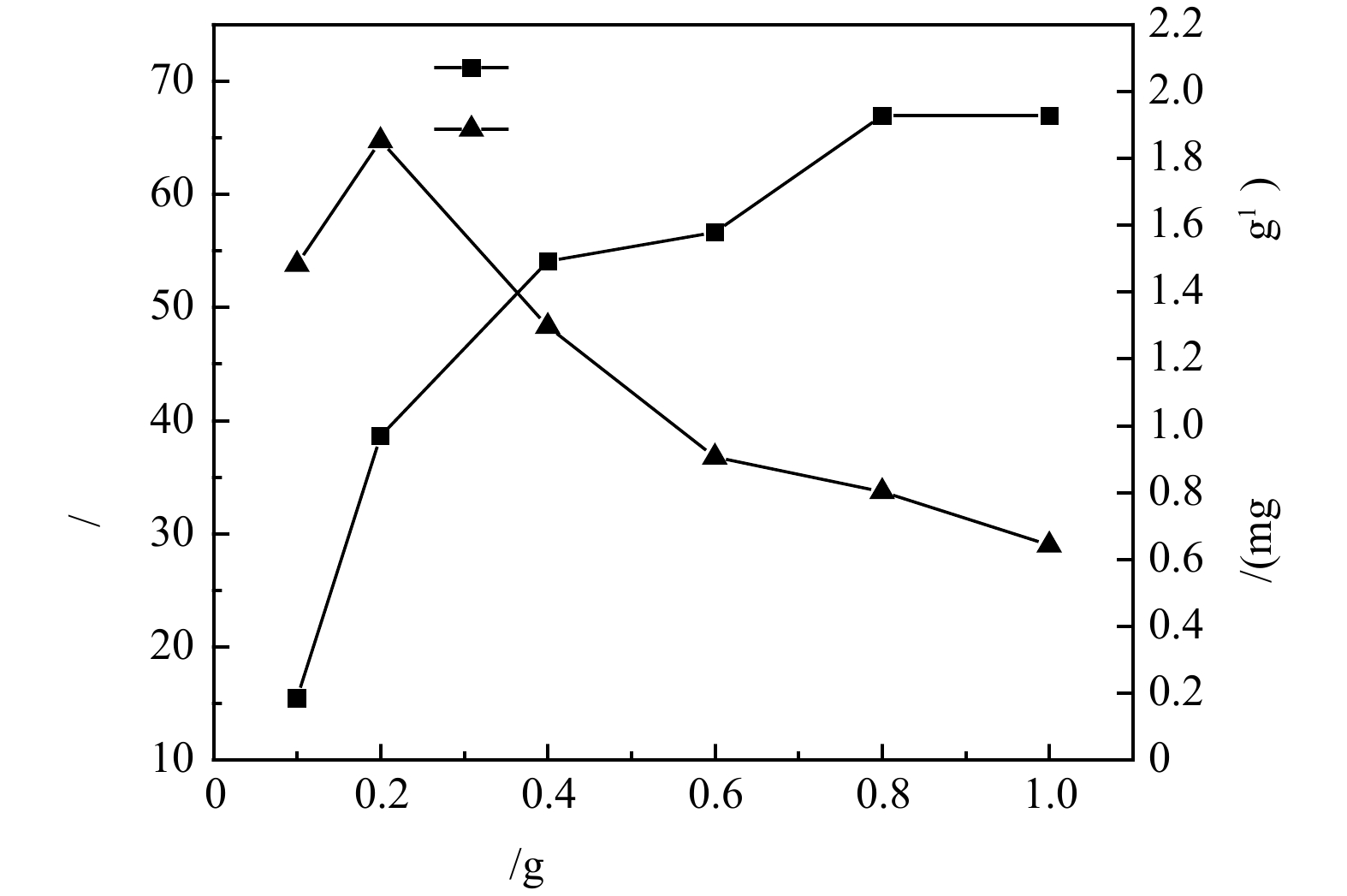 图 2 氨氮去除率和吸附量随沸石投加量的变化规律Figure 2. Variations of the removal rate and adsorption capacity of ammonia with zeolite dosage
图 2 氨氮去除率和吸附量随沸石投加量的变化规律Figure 2. Variations of the removal rate and adsorption capacity of ammonia with zeolite dosage2)初始氨氮浓度对沸石脱氨效果的影响。沸石对氨氮的去除率及吸附量随初始氨氮浓度变化规律如图3 所示。可见,沸石对氨氮的去除率随氨氮初始浓度的增加而下降,而吸附量却随之增加,当沸石投加量为4 g·L−1,初始氨氮质量浓度为100 mg·L−1时,沸石对氨氮吸附量可提升到3.89 mg·g−1,但氨氮去除率也降至17%。随着氨氮浓度的增加,体系中的
NH+4 NH+4  图 3 氨氮去除率和吸附量随氨氮初始浓度的变化规律Figure 3. Variations of the removal rate and adsorption capacity of ammonia with initial ammonia concentration.
图 3 氨氮去除率和吸附量随氨氮初始浓度的变化规律Figure 3. Variations of the removal rate and adsorption capacity of ammonia with initial ammonia concentration.3)沸石对氨氮的吸附动力学研究。采用准一级和准二级动力学模型,对沸石投加量为4 g·L−1,初始氨氮质量浓度为10 mg·L−1的24 h吸附动力学实验数据进行拟合分析。由图4可以看出,在吸附开始的1 h内,沸石对氨氮吸附速率较快,1 h后吸附量缓慢上升,12 h吸附趋于平衡,平衡吸附量在1.3 mg·g−1左右。在吸附初期,沸石颗粒上吸附位点较多,氨氮浓度高,吸附较快,而随着吸附进行,氨氮浓度降低,吸附位点被占用,吸附速率下降。此外,沸石的表面物理吸附及
NH+4 NH+4 qt t 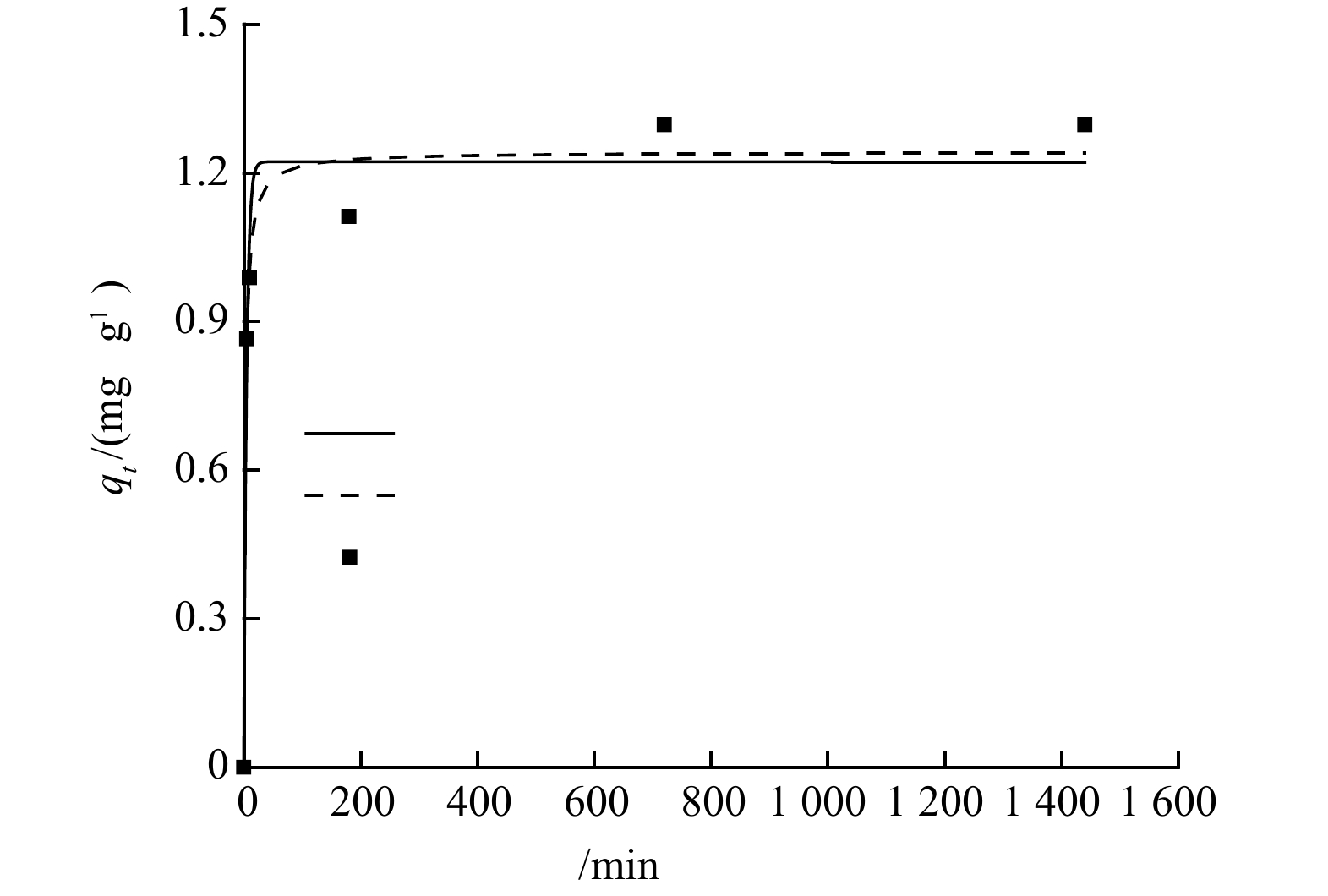 图 4 沸石吸附氨氮的准一级与准二级动力学模型拟合曲线Figure 4. Pseudo-first-order model and pseudo-second-order model curves for ammonia adsorption onto zeolite表 1 准一级与准二级动力学方程拟合参数及相关系数Table 1. Fitting parameters and correlation coefficients of pseudo-first order kinetics and pseudo second order kinetics
图 4 沸石吸附氨氮的准一级与准二级动力学模型拟合曲线Figure 4. Pseudo-first-order model and pseudo-second-order model curves for ammonia adsorption onto zeolite表 1 准一级与准二级动力学方程拟合参数及相关系数Table 1. Fitting parameters and correlation coefficients of pseudo-first order kinetics and pseudo second order kineticsqe,exp /(mg·g−1) 准一级动力学 准二级动力学 qe,celc/(mg·g−1) k1/min−1 R2 qe,celc/(mg·g−1) k2/(g·(mg·min)−1) R2 1.30 1.22 0.21 0.969 1.24 0.34 0.982 | Show Table DownLoad:
CSV
DownLoad:
CSV
4)沸石对氨氮的吸附等温线。在25 ℃的条件下,测定4 g·L−1沸石在不同初始氨氮浓度下的平衡吸附量,进行吸附等温研究,结果见图5。吸附等温模型是用于描述在特定实验温度下,吸附剂与吸附质之间的相互作用[13]。本研究使用Langmuir和Freundlich模型(表2) 对数据进行分析,以准确地研究氨氮在沸石上的吸附机理。首先分别以
1/qe 1/Ce lgqe lgCe n > 1 0.1 < 1/n < 0.5 1/n 1/n > 2 1/n 表 2 Langmuir与Freundlich吸附等温模型拟合参数及相关系数Table 2. Fitting parameters and correlation coefficients of adsorption isotherm models of Langmuir and Freundlich温度/ ℃ Langmuir Freundlich qm/(mg·g−1) b/(L·mg−1) R2 Kf/(mg(1−1/n) g−1 L1/n) 1/n R2 25 4.12 0.33 0.995 0.69 0.40 0.960 | Show TableDownLoad:
CSV
2.2 沸石-动态膜系统脱氨效能和成膜特征研究
1) 沸石动态膜系统脱氨效果。在初始氨氮质量浓度10 mg·L−1,2 L体系,电动搅拌器室温搅拌1 h条件下,吸附动态膜系统中沸石对氨氮的去除率及吸附量随沸石投加量的变化规律见图6。由图6可以看出,与在100 mL体系中、25 ℃、180 r min−1恒温振荡箱和初始氨氮质量浓度10 mg·L−1反应条件(图2)相比,该条件下沸石对氨氮的去除率与吸附量稍有降低,基本能够充分发挥沸石的吸附脱氨效能,不会造成沸石动态膜系统内沸石吸附位点的浪费。当沸石投加量达到10 g·L−1以后,对氨氮去除率的提升非常有限,基本在55%~60%,吸附量随沸石投加量的增加逐渐下降。为了保证吸附效果,循环吸附-动态膜系统中沸石的投加量以10 g·L−1为宜,此时的去除率与吸附量分别为56%和0.59 mg·g−1。本系统中,沸石吸附容量与NaCl改性沸石[15]吸附容量(0.56 mg·g−1,初始氨氮质量浓度20 mg·L−1,投加量30 g·L−1,室温30 ℃)较为接近,但氨氮去除率明显低于改性4A沸石分子筛(91%)[14] 和NaCl改性沸石(83%)[15],氨氮去除率的差距可能是由于沸石投加量增大和初始浓度差异引起的,后续沸石动态膜系统可根据实际脱氨需要进行沸石改性优化。
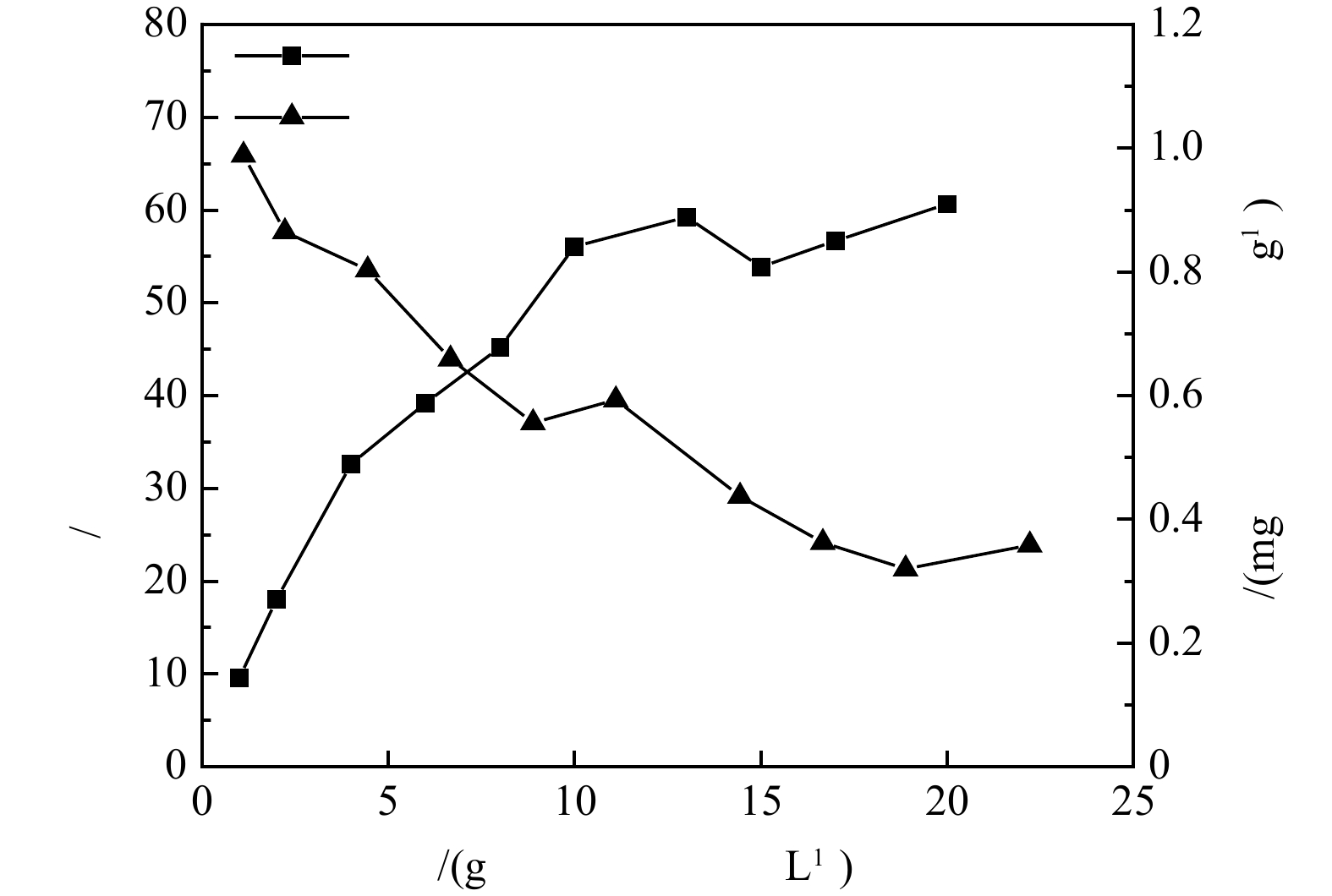 图 6 沸石-动态膜系统中氨氮去除率及吸附量随沸石投加量的变化规律Figure 6. Variations of ammonia nitrogen removal rate and adsorption amount with zeolite dosage in the zeolite-dynamic membrane system
图 6 沸石-动态膜系统中氨氮去除率及吸附量随沸石投加量的变化规律Figure 6. Variations of ammonia nitrogen removal rate and adsorption amount with zeolite dosage in the zeolite-dynamic membrane system2)沸石动态膜系统的成膜特征。结果表明,在沸石粉投加量为1 g·L−1,蠕动泵流量为40 mL·min−1和38 μm支撑膜的条件下,沸石动态膜的形成效果最佳,其浊度与跨膜压的变化规律如图7(a)所示。从图7(a)中可知,动态膜出水浊度在 5 min即从最初的145.5 NTU降低到25.3 NTU,10 min时降至7.52 NTU,此时跨膜压开始上升,出水浊度也开始缓慢波动上升。动态膜整体形成过程维持了约45 min,而跨膜压在40 min内上升至约95 kPa。在38 μm支撑膜截留作用下,沸石动态膜系统出水浊度逐渐下降,随着动态膜层不断密实,沸石动态膜对小粒径沸石截留效果也在逐渐提升,但实验观察到出水浊度经过下降阶段后,随压力的上升呈现出不断缓慢上升趋势。推测可能是在动态膜形成过程中,进水的冲击及沸石颗粒性质造成动态膜滤饼层并不稳定,在跨膜压作用下小颗粒沸石穿透膜层导致浊度升高。实验中发现在此条件下沸石形成的动态膜层较薄,不太均匀(图7(b)),说明沸石动态膜的稳定性有待进一步提升。若以浊度值去除率近似估算沸石回收率,沸石动态膜系统对沸石颗粒的回收率最高可达到94.8%。
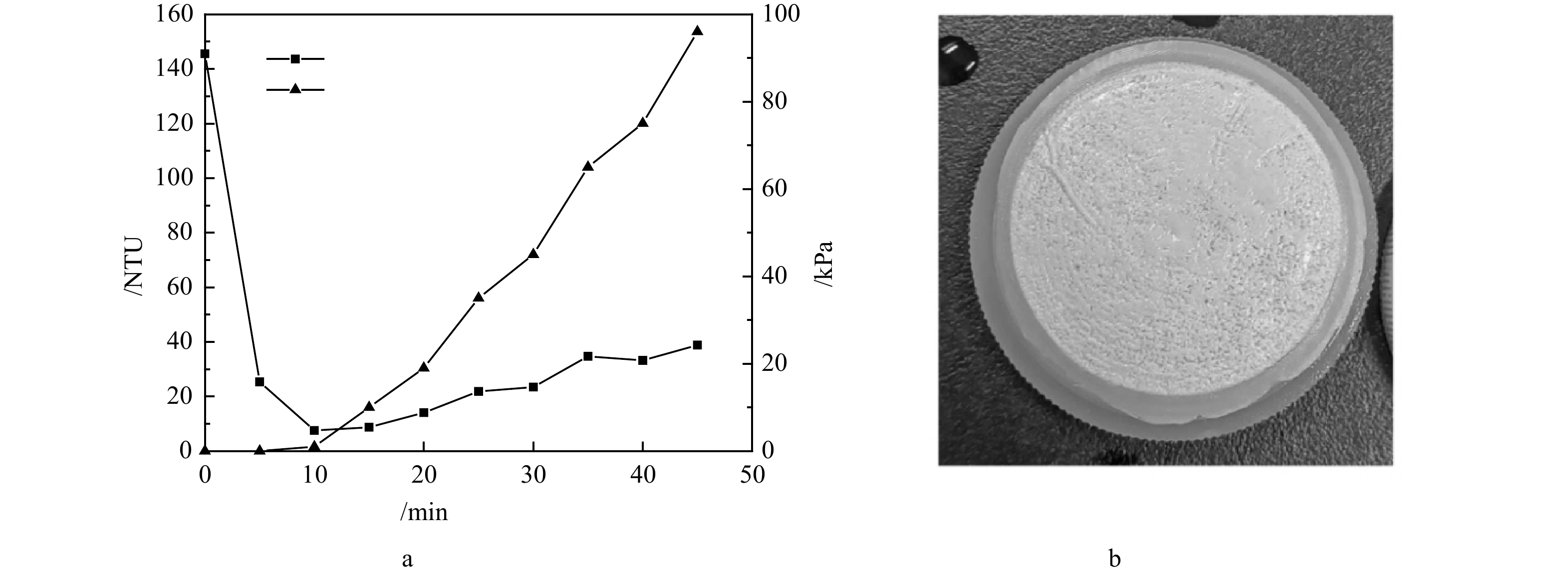 图 7 沸石动态膜成膜过程中出水浊度与跨膜压的变化规律和成膜图像Figure 7. Changes in effluent turbidity and transmembrane pressure during the zeolite DM formation and DM picture
图 7 沸石动态膜成膜过程中出水浊度与跨膜压的变化规律和成膜图像Figure 7. Changes in effluent turbidity and transmembrane pressure during the zeolite DM formation and DM picture3)复合硅藻土沸石动态膜系统成膜特征和脱氨效果。为了有效提升沸石动态膜稳定性,同时保证动态膜对氨氮的去除效果,后续实验选用硅藻土作为辅助成膜材料,以1:1质量比与沸石进行复配。为了避免动态膜形成过快增加动态膜形成过程的监测困难,先投加10 g·L−1沸石与10 g·L−1硅藻土于2 L 10 mg·L−1氨氮废水中,搅拌1 h以充分混合,然后取200 mL混合液稀释10倍后开始构建复合硅藻土沸石动态膜,在动态膜形成过程中出水浊度与跨膜压随时间的变化情况如图8(a)所示。沸石与硅藻土按质量比1:1复配形成动态膜时,出水浊度下降很快,5 min即由81.6 NTU下降至1.23 NTU,10 min后一直稳定在1 NTU以下,表明动态膜已稳定形成,且连续运行55 min直至跨膜压上升至82 kPa。硅藻土的加入使出水浊度的稳定性得到明显改善,相比于沸石动态膜,沸石与硅藻土混合复配吸附动态膜形成快速且稳定,实验结束后发现复合硅藻土沸石动态膜更加致密而均匀(图8(b)),可以有效截留微尺度沸石。从跨膜压的变化规律来看,沸石与硅藻土复配也一定程度上延长了装置的运行时间。因此,沸石与硅藻土复配形成的动态膜可以有效回收微尺度沸石。若以浊度值去除率近似估算硅藻土和沸石的回收率,复合硅藻土沸石动态膜系统回收率可达到98.77%,高于动态膜对微尺度改性活性炭的回收率(63%~87%)[16]。
 图 8 复合硅藻土沸石成膜过程中出水浊度与跨膜压的变化规律及成膜图像Figure 8. Variations of effluent turbidity and TMP during the DM formation with zeolite and diatomite and DM picture
图 8 复合硅藻土沸石成膜过程中出水浊度与跨膜压的变化规律及成膜图像Figure 8. Variations of effluent turbidity and TMP during the DM formation with zeolite and diatomite and DM picture沸石与硅藻土混合复配成膜过程中氨氮浓度的变化规律如图9所示。由图9可知,在反应开始时,在初始氨氮质量浓度为10 mg·L−1时,经过60 min后剩余氨氮质量浓度为4.4 mg·L−1,去除率可达56%,与单独投加沸石时对氨氮的去除效果相近。硅藻土本身对氨氮的吸附效果不佳,该吸附过程主要还是以沸石吸附为主。60 min稀释后开始形成动态膜时,系统内与出水的氨氮质量浓度(折算为稀释前)在6.5 mg·L−1左右,比稀释前略有提升,可能是因为稀释过程导致了氨氮吸附平衡的移动,沸石吸附的氨氮部分转移到了稀释后溶液中。由复合硅藻土沸石动态膜出水浊度的变化规律可知,系统运行5 min即可形成稳定的动态膜,随着动态膜的持续形成和过滤,动态膜出水氨氮浓度略低于反应系统内氨氮浓度。这可能是因为在动态膜成膜前系统混合搅拌过程中,沸石对氨氮的吸附已基本趋于平衡,而氨氮废水经过动态膜时与沸石接触时间较短,无法进行充分接触,因此,复合硅藻土沸石动态膜层对氨氮的去除效果有限。复合硅藻土-沸石-动态膜系统实现了废水中氨氮的吸附去除,同时回收了微颗粒沸石,可为吸附材料的再生和回收利用提供参考。
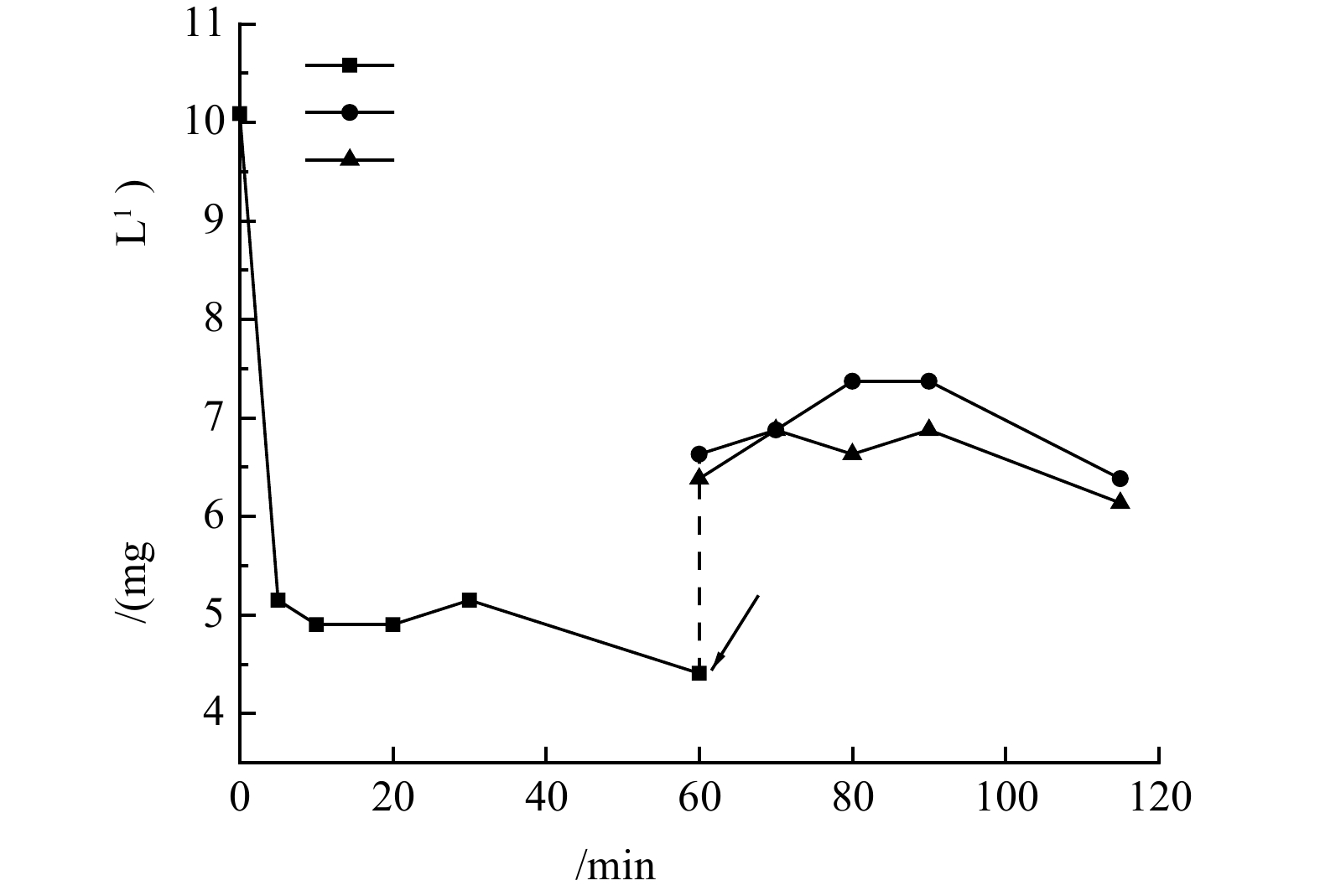 图 9 复合硅藻土沸石动态膜形成过程中氨氮浓度的变化规律Figure 9. Variation of ammonia nitrogen concentration during the DM formation with zeolite and diatomite
图 9 复合硅藻土沸石动态膜形成过程中氨氮浓度的变化规律Figure 9. Variation of ammonia nitrogen concentration during the DM formation with zeolite and diatomite整体来看,沸石静态吸附与沸石动态膜系统在氨氮去除性能上的表现相差不大,但沸石动态膜系统的优势在于可以对沸石吸附颗粒进行有效回收。尽管单纯沸石构建动态膜系统出现了出水浊度波动上升和沸石颗粒穿透,但通过复合硅藻土沸石动态膜系统能够保证了沸石颗粒的有效回收,从而提升了动态膜系统稳定性,也同步实现了废水中氨氮的去除。
3. 结论
1)在沸石静态吸附研究中,在初始氨氮质量浓度为10 mg·L−1,沸石投加量为4 g·L−1时,沸石对氨氮去除率和吸附量分别可达54%和1.3 mg·g−1;当初始氨氮质量浓度增至100 mg·L−1时,沸石对氨氮吸附量可提升至3.89 mg·g−1,但氨氮去除率也降至17%。
2)沸石脱氨过程更符合准二级动力学过程;Langmuir吸附等温模型对沸石吸附脱氨拟合效果更优,理论最大平衡吸附量为4.12 mg·g−1。
3)在10 g·L−1沸石投加量下,沸石动态膜系统对氨氮的去除率为56%,出水浊度可降至7.52 NTU,沸石动态膜的稳定性有待进一步提升。
4)沸石与硅藻土按照1:1的质量比复配,10 min即可快速构建复合硅藻土沸石动态膜,出水浊度稳定在1 NTU以下,氨氮去除率可达到56%,在脱氨的同时能够实现沸石的有效回收,并显著提升了复合硅藻土沸石动态膜系统的稳定性。沸石吸附是复合沸石动态膜系统脱氨的主要作用机制。
-
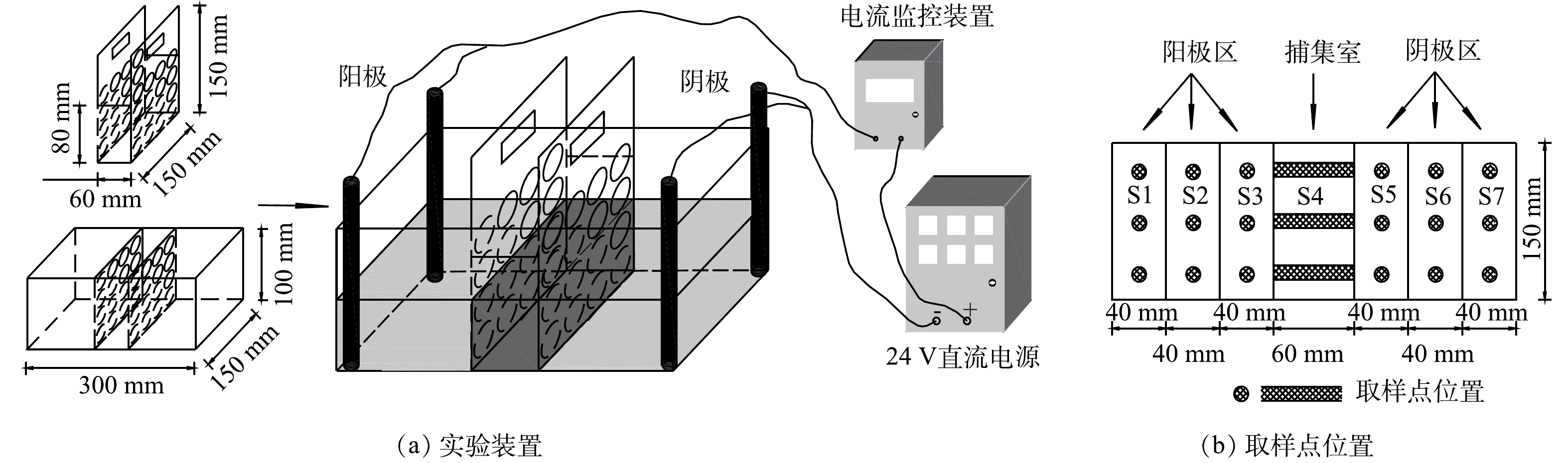
图 1 实验装置及取样点位置
Figure 1. Schematic diagram of experimental setup and sampling positions
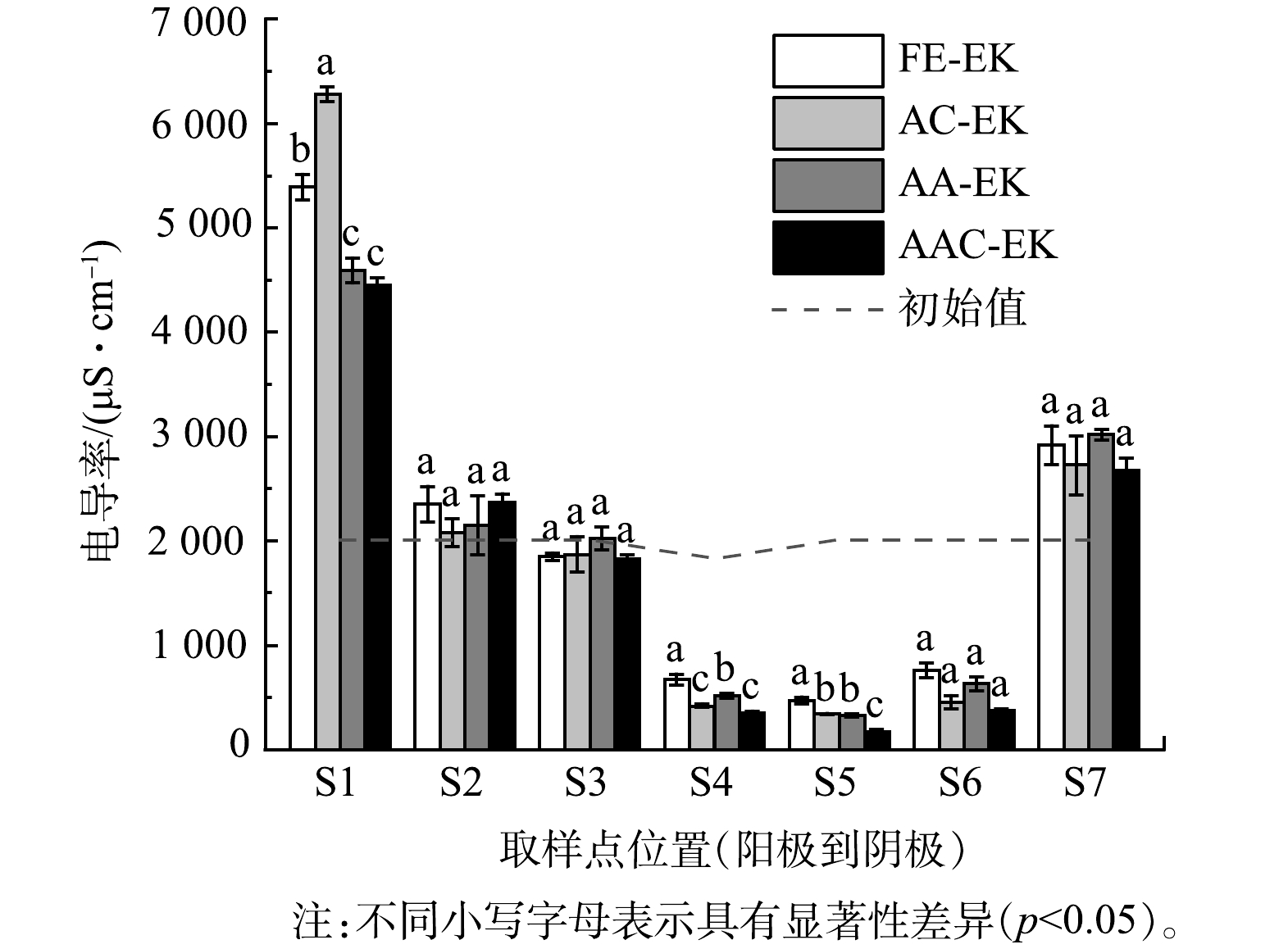
图 3 电动修复后土壤电导率分布
Figure 3. Distribution of conductivity in soil after electrokinetic treatment
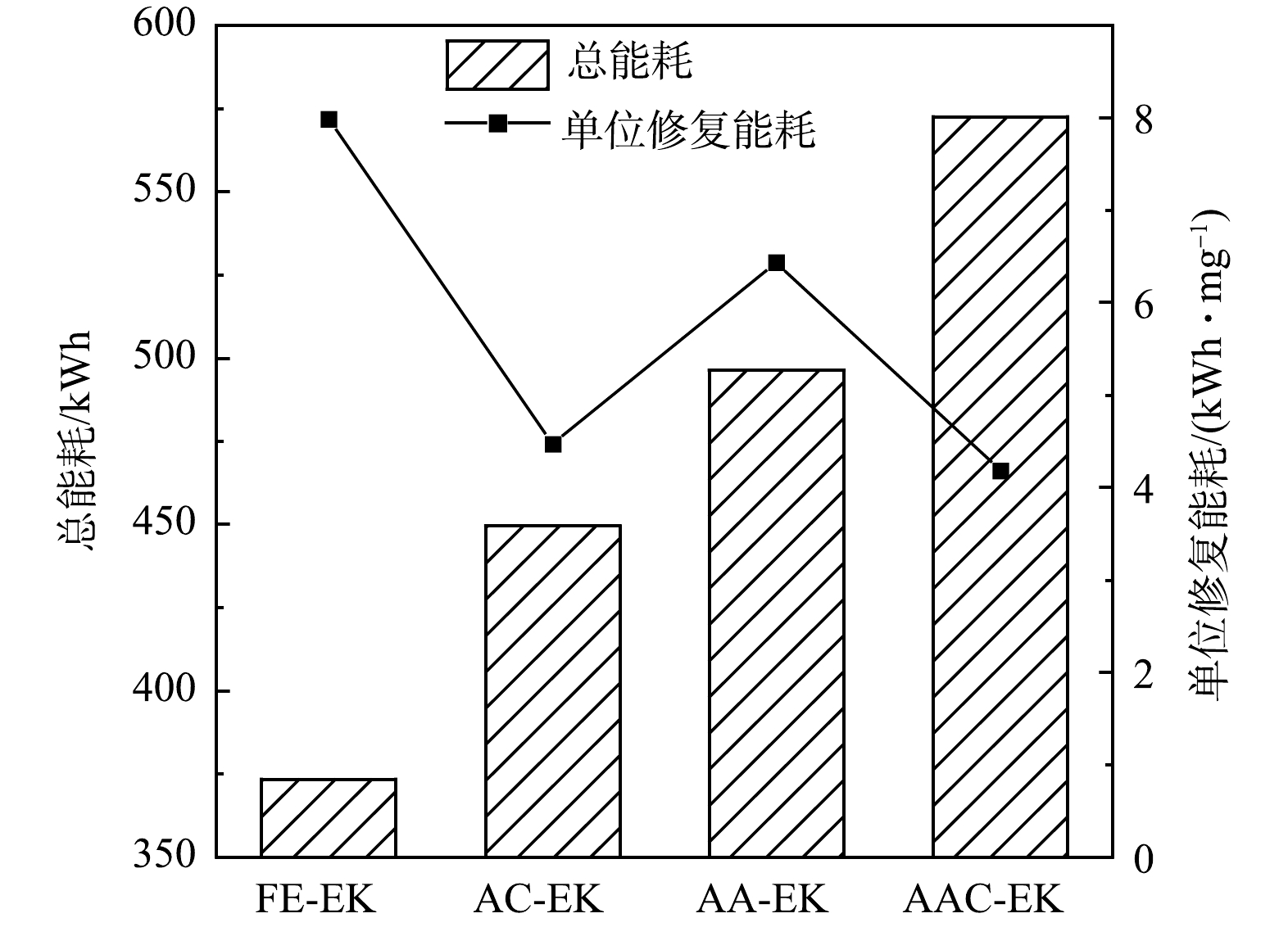
图 7 不同电动处理组总能耗及单位修复能耗
Figure 7. Total energy consumption and energy consumption per unit of remediation of different electrokinetic treatments

图 8 电动结束后土壤中As(V)和As(III)分布
Figure 8. Distributions of As(V) and As(III) in soil after electrokinetic treatment
-
[1] 赵宇, 艾雯妍, 文思颖, 等. 微生物-植物联合修复镉砷污染农田土壤技术与应用[J]. 生态毒理学报, 2022, 17(6): 144-162. [2] LI J P, DING Y, WANG K L, et al. Comparison of humic and fulvic acid on remediation of arsenic contaminated soil by electrokinetic technology[J]. Chemosphere, 2020, 241: 125038. doi: 10.1016/j.chemosphere.2019.125038 [3] 吕紫娟, 王华伟, 吴雅静, 等. 纳米零价铁物相转变对砷污染土壤稳定化效果和潜在毒性的影响[J]. 环境工程, 2022, 40(3): 24-31. [4] 骆永明, 滕应. 我国土壤污染的区域差异与分区治理修复策略[J]. 中国科学院院刊, 2018, 33(2): 145-152. doi: 10.16418/j.issn.1000-3045.2018.02.003 [5] ALKA S, SHAHIR S, IBRAHIM N, et al. Arsenic removal technologies and future trends: A mini review[J]. Journal of Cleaner Production, 2021, 278: 123805. doi: 10.1016/j.jclepro.2020.123805 [6] MA C Z, LI J P, XIA W, et al. Effect of additives on the remediation of arsenic and chromium co-contaminated soil by an electrokinetic-permeable reactive barrier[J]. Environmental Science and Pollution Research, 2022, 29(8): 11966-11975. doi: 10.1007/s11356-021-16357-1 [7] XU Y F, LU Q Q, LI J P, et al. Effect of humus on the remediation of arsenic-contaminated soil by electrokinetic technology[J]. Environmental Technology & Innovation, 2021, 21(14): 101297. [8] KARACA O, CAMESELLE C, BOZCU M. Opportunities of electrokinetics for the remediation of mining sites in Biga peninsula, Turkey[J]. Chemosphere, 2019, 227: 606-613. doi: 10.1016/j.chemosphere.2019.04.059 [9] YAO W K, CAI Z P, SUN S Y, et al. Electrokinetic-enhanced remediation of actual arsenic-contaminated soils with approaching cathode and Fe0 permeable reactive barrier[J]. Journal of Soils and Sediments, 2020, 20(3): 1526-1533. doi: 10.1007/s11368-019-02459-4 [10] 付博, 王刚, 张志彬, 等. pH与Eh对郑州北郊水源地沉积物中砷溶出的影响[J]. 青岛理工大学学报, 2013, 34(4): 99-103. [11] 周一敏, 黄雅媛, 刘凯, 等. 典型铁、锰矿物对稻田土壤砷形态与酶活性的影响[J]. 环境科学, 2022, 43(5): 2732-2740. [12] JI D L, ZHANG J, MENG F S, et al. Species and distribution of arsenic in soil after remediation by electrokinetics coupled with permeable reactive barrier[J]. Water, Air, & Soil Pollution, 2020, 231(12): 567. [13] 中华人民共和国生态环境部. 土壤环境质量建设用地土壤污染风险管控标准(试行): GB 36600—2018[S]. 北京: 中国环境科学出版社, 2018. [14] 尹静玄, 王平, 徐海音, 等. 耐镉细菌联合电动技术修复镉污染土壤的研究[J]. 环境科学学报, 2020, 40(6): 2212-2219. [15] 中华人民共和国生态环境部. 土壤氧化还原电位的测定 电位法: HJ 746—2015[S]. 北京: 中国环境科学出版社, 2015. [16] 刘向磊, 孙文军, 文田耀, 等. 三酸分步消解-电感耦合等离子体质谱法测定土壤详查样品中23种金属元素[J]. 岩矿测试, 2020, 39(5): 793-800. [17] ZHENG J, HINTELMANN H, DIMOCK B, et al. Speciation of arsenic in water, sediment, and plants of the Moira watershed, Canada, using HPLC coupled to high resolution ICP–MS[J]. Analytical and Bioanalytical Chemistry, 2003, 377(1): 14-24. doi: 10.1007/s00216-003-1920-3 [18] 张静, 刘晓端, 江林. 土壤中不同形态砷的分析方法[J]. 岩矿测试, 2008(3): 179-183. doi: 10.3969/j.issn.0254-5357.2008.03.005 [19] 黄中情, 杨常亮, 张璟, 等. 碳酸氢盐对沉积物中砷迁移转化的影响[J]. 环境科学与技术, 2020, 43(11): 69-75. doi: 10.19672/j.cnki.1003-6504.2020.11.009 [20] 孟欣, 李刚, 高鹏, 等. 高羊茅对电动-微生物修复石油污染土壤的影响[J]. 农业环境科学学报, 2020, 39(7): 1532-1539. doi: 10.11654/jaes.2019-1438 [21] BESSAIM M M, KARACA O, MISSOUM H, et al. Effect of imposed electrical gradient on removal of toxic salt contaminants from alkali-saline low permeable soil during electrokinetic remediation[J]. Arabian Journal of Geosciences, 2020, 13(14): 1-12. [22] XU H T, CANG L, SONG Y, et al. Influence of electrode configuration on electrokinetic-enhanced persulfate oxidation remediation of PAH-contaminated soil[J]. Environmental Science and Pollution Research, 2020, 27(35): 44355-44367. doi: 10.1007/s11356-020-10338-6 [23] SHEN Z M, ZHANG J D, QU L Y, et al. A modified EK method with an I−/I2 lixiviant assisted and approaching cathodes to remedy mercury contaminated field soils[J]. Environmental Geology, 2009, 57(6): 1399-1407. doi: 10.1007/s00254-008-1418-6 [24] 周丽玮, 王航, 刘阳生. 转换电极的电动力强化植物修复高浓度砷污染土壤[J]. 环境工程, 2020, 38(10): 228-233. doi: 10.13205/j.hjgc.202010036 [25] SHIN S Y, PARK S M, BAEK K. Electrokinetic removal of As from soil washing residue[J]. Water, Air, & Soil Pollution, 2016, 227(7): 223. [26] 周实际, 杜延军, 倪浩, 等. 压实度对铁盐稳定化砷、锑污染土特性的影响及机制研究[J]. 岩土力学, 2022, 43(2): 432-442. doi: 10.16285/j.rsm.2021.1474 [27] RYU S R, JEON E K, BAEK K. A combination of reducing and chelating agents for electrolyte conditioning in electrokinetic remediation of As-contaminated soil[J]. Journal of the Taiwan Institute of Chemical Engineers, 2017, 70: 252-259. doi: 10.1016/j.jtice.2016.10.058 [28] 胡立琼, 曾敏, 雷鸣, 等. 含铁材料对污染水稻土中砷的稳定化效果[J]. 环境工程学报, 2014, 8(4): 1599-1604. [29] 蒋毅, 刘雅, 辜娇峰, 等. 三元复合调理剂对土壤镉砷赋存形态和糙米镉砷累积的调控效应[J]. 环境科学, 2021, 42(1): 378-385. doi: 10.13227/j.hjkx.202006126 [30] 邓天天, 胡烨, 刘帅霞, 等. Fe2O3@GO聚合物对水中As3+的吸附特性表征[J]. 生态与农村环境学报, 2018, 34(10): 930-938. [31] 蒋成爱, 吴启堂, 陈杖榴. 土壤中砷污染研究进展[J]. 土壤, 2004, 36(3): 264-270. doi: 10.13758/j.cnki.tr.2004.03.007 期刊类型引用(11)
1. 王鑫,张彪,廖述聪,吴小缓,张体良. 氨碱白泥综合利用研究进展. 水泥. 2025(03): 79-82 .  百度学术
百度学术
2. 陈永亮,成亮,陈铁军,陈君宝,张轶轲,夏加庚. 砖混建筑垃圾制备蒸压加气混凝土性能及水化机理. 材料导报. 2024(12): 126-131 . 百度学术
3. 孙鑫蕊,王学志,辛明,贺晶晶. 硅锰渣复合粉煤灰制备免蒸压加气混凝土. 金属矿山. 2023(01): 289-295 . 百度学术
4. 陈潇,张浩宇,薛鑫,杨寅,龚天天,张刘阳. 固体废弃物在蒸压加气混凝土中的应用现状综述. 硅酸盐通报. 2023(02): 541-553 . 百度学术
5. 房天齐,黄舒,乔秀臣. 循环流化床灰蒸压加气混凝土中延迟形成钙矾石的研究. 硅酸盐通报. 2023(07): 2439-2446 . 百度学术
6. 余海燕,胡林童. 轻质碱渣-氯氧镁水泥基材料性能研究及孔结构特征分析. 混凝土与水泥制品. 2022(04): 78-83 . 百度学术
7. 傅峰. 蒸压加气混凝土砌块的制备及抗压性能研究. 菏泽学院学报. 2021(02): 66-72 . 百度学术
8. 迂晓轩,彭孟啟,齐贺,高昊元,丁艳玲,梁琨,王静贻,邢芸,鲁官友,周鼎. 建筑工程垃圾减量化概况及评价标准探究. 环境工程. 2020(03): 1-8 . 百度学术
9. 顾城名,刘品德,夏理想,施星宇,余甲锋. 固体废弃物在蒸压加气混凝土中的利用与研究进展. 砖瓦. 2020(05): 64-68 . 百度学术
10. 顾城名,刘品德,夏理想,施星宇,余甲锋. 固体废弃物在蒸压加气混凝土中的利用与研究进展. 砖瓦. 2020(06): 82-85 . 百度学术
11. 徐东,倪文,汪群慧,许成文,姜瑶琪. 碱渣复合胶凝材料制备无熟料混凝土. 哈尔滨工业大学学报. 2020(08): 151-160 . 百度学术
其他类型引用(17)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2526
- HTML全文浏览数: 2526
- PDF下载数: 127
- 施引文献: 28
