



-
氮、磷污染是造成水体富营养化的主要原因,同时氮和磷也是一种重要资源,是生产农肥和畜禽饲料的原料。全球磷资源十分有限,且单向流动、难以再生。在现有技术、经济水平条件下,全球探明可供开采的磷矿资源不足100 a[1]。富含氮磷元素废水的排放不仅会造成氮磷的流失,还会造成地表水体富营养化。因此,高效回收废水中的氮磷资源,已成为相关领域的研究热点之一。
目前的氮磷共存水体主要是养殖废水、食品废水和市政污水。其中山东某食品公司产生的废水氨氮和总磷含量高达160 mg·L−1和100 mg·L−1,经过初沉池、UASB池、A/O池和二沉池处理后氨氮含量显著降低,但总磷含量依然高达77 mg·L−1[2]。此外,在甘肃某养牛场养殖废水中氨氮和磷酸盐质量浓度也高达1 304 mg·L−1和146 mg·L−1[3]。目前,最常用的脱氮除磷工艺为氧化沟工艺、A2O工艺和SBR工艺。但上述工艺对污水水质的稳定性要求较高,水质的大幅度变化会影响微生物脱氮除磷效果。此外,污水经二级处理后增加深度处理方能较好净化水质,但随之而来的是高昂的化学添加剂成本以及基建和电力费用。因此,寻找一种廉价的清洁材料用于同步、高效去除氮磷对于治理氮磷废水至关重要。
目前,磷酸铵镁(magnesium ammonium phosphate,MAP)结晶法往往具备同步实现对氮、磷的高效脱除和有效回收,广泛用于尿液、垃圾渗滤液、焦化废水的处理[4-6]。MAP英文俗名为struvite,中文俗称鸟粪石,化学成分为MgNH4PO4·6H2O,是一种较难溶于水的无色、白色(脱水后)、黄色、棕色或浅灰色的晶体,且含有氮、磷植物营养元素,是一种很好的缓释化肥[7-8]。其基本原理是在沼液中投加Mg2+,在碱性条件下使沼液中的PO43−、HPO42−、H2PO4−及NH4+与Mg2+反应成生MgNH4PO4·6H2O(struvite,即鸟粪石),从而回收水体的氮磷[9]。传统的MAP结晶法回收率较低,如磷结晶率达90%以上时,氨氮结晶率仅为13%左右,难以达到两者同步高效结晶[10]。鸟粪石的形成需要在较高的溶液pH条件下,需投加含Mg2+沉淀剂和碱溶液,原材料成本过高限制该方法的使用,且反应过后的材料回收困难[11]。HUANG等[12]利用镁盐改性天然沸石同步去除废水中的氮磷获得良好的去除效果。针对以上问题,选择一种自身碱性高、成本低和易回收的材料尤为重要。
常见镁来源有Mg(OH)2、MgSO4、MgCl2等。其中,MgO由于其安全稳定且自身碱性高等优点,受到广泛应用[10, 12-13]。生物炭比表面积大、离子交换能力强,可以去除水中的污染物[13-14]。并且生物炭表面呈负电荷,可以吸附水中的氨氮,但通常不能吸附磷酸盐。利用金属氧化物进行生物炭进行改性,可以有效改善其对磷的吸附性能[14]。生物质原料包括农业和森林残余物及其副产品(水稻秸秆[15]、小麦秸秆[16]、玉米芯[17]和菜叶[18]等)、动物粪便、造纸厂废料、城市固体废物和污泥[19-20]。选择市政污泥作为原料制备污泥基生物炭,具有来源稳定、成本低等优点,同时也为污泥的资源化利用提供了一种途径,具有良好社会意义。
因此,本研究开发了一种以镁离子为靶向供给、碱性调控能力强、成本低廉且易于回收的新型磁性污泥基生物炭复合材料(Mg/Fe sludge biochar, MF-SBC),用于废水中氮磷的同步回收。考察了初始pH、接触时间、共存离子、投加量对氮磷回收效果的影响,采用动力学模型等分析了回收特征,并结合XRD、XPS、SEM等多种表征手段探讨了新型磁性污泥基生物炭对水溶液中氮磷同步回收的作用机制,为其实际应用提供参考。
-
实验所用试剂包括七水合硫酸亚铁(FeSO4·7H2O)、六水合氯化镁(MgCl2·6H2O)、氢氧化钠(NaOH)、氯化铵(NH4Cl)和磷酸二氢钾(KH2PO4),以上试剂均为化学纯或分析纯。使用去离子水制备所有溶液。本实验原料取自中国江苏省武进区武南污水厂的脱水污泥,将脱水污泥在105 ℃条件下烘至恒重,研磨过200目筛,置于干燥器中备用。通过X射线荧光光谱仪检测到污泥粉末中主要的无机化学成分有SiO2、Al2O3、CaO、MgO、Fe2O3和P2O5。
-
MF-SBC的制备具体步骤如下:准确称取2.5 g干燥的污泥粉末加入到50 mL 0.2 mmol·L−1 FeSO4·7H2O溶液中浸泡并振荡6 h;将污泥/FeSO4混合溶液放入烘箱中经80 ℃烘干并研磨过200目筛,之后将其加入50 mL 1.25 mmol·L−1 MgCl2·6H2O溶液,并进行磁力搅拌30 min;随后,向上一步得到的混合溶液中缓慢加入50 mL 2.5 mmol·L−1 NaOH溶液并磁力搅拌12 h,紧接着陈化24 h;过滤以上溶液后所得沉淀物用去离子水反复清洗至中性,再经80 ℃烘干;将得到的干燥沉淀物放置管式炉中以5 ℃·min−1慢速升温至450 ℃,并在该温度下持续煅烧2 h,得到磁性MF-SBC。
-
分别使用KH2PO4和NH4Cl配置磷酸盐和氨氮储备溶液,并稀释至实验所需浓度。对于每批次实验,将一定量的MF-SBC材料加入到100 mL含有磷酸盐和氨氮的锥形瓶中,并用0.1 mmol·L−1 HCl和NaOH调节溶液至所需pH,之后将密封的锥形瓶置于恒温振荡器中振荡。反应结束后,取上层清液,用0.45 μm滤膜过滤后用UV-vis分光光度计(pharo300,merck, 德国)检测。分别考察了初始pH、氨氮和磷酸盐的质量浓度、MF-SBC投加量和反应时间对MF-SBC同步吸附氨氮和磷酸盐的影响,各批次实验的详细实验条件见表1。
-
1)生物炭表征。采用阶级开门扫描方式进行分析,扫描角2θ范围为10°~80°,工作电压和电流分别为60 kV和300 mA;分别利用扫描电子显微镜(SEM,SUPRA5,德国)和快速全自动比表面和孔径分析仪(BET,Autosorb-iQ2-MP,美国)来观察和测定微观形貌和比表面积;使用X射线光电子能谱仪(XPS,EscaLab 250Xi,美国)和能量色散X射线光谱(EDX)分析样品结构表面处的局部元素组成。在400~4 000 cm−1内通过傅里叶光谱仪(FTIR,Nicolet IS5 美国)记录红外光谱,鉴定所制备样品官能团。
2)溶液中污染物浓度的测定。采用国标钼酸铵分光光度法(GB11893-89)和纳氏试剂分光光度法(GB11893-89)测定溶液中磷和氨氮含量,每组3个平行样,到达反应平衡时,利用式(1)计算MF-SBC的去除量。
式中:Qe为吸附剂的吸附量,mg·g−1;C0为溶液的初始质量浓度,g·L−1;Ce为吸附后的溶液质量浓度,mg·L−1;V为溶液的体积,L;M为吸附剂质量,g。
-
为分析MF-SBC对磷酸盐和氨氮的吸附动力学情况,采用准一级动力学方程(式(2))、准二级动力学方程(式(3))和颗粒内扩散方程(式(4))对其进行拟合。
式中:t表示吸附时间,min;Qt表示t时吸附剂的吸附量,mg·g−1;Qe表示吸附平衡的吸附量,mg·g−1;k1为一级速率常数,min−1;k2为二级速率常数,g·(mg·min)−1;kp表示颗粒内扩散速率常数,mg·(mg·min)−1;C是常数,为颗粒内扩散方程的截距。
-
吸附实验结束后,过滤收集MF-SBC,用1 mol·L−1 NaOH溶液超声30 min并静置浸渍1 h,再用去离子水冲洗至pH为中性后烘干,干燥保存并用于下一次的循环使用。
-
为了确定MF-SBC的磁性,在室温下通过振动样品磁强计(VSM)来测量材料的磁性,得到磁滞回线(图1(a))。由图1(a)可以看出,MF-SBC的曲线显示出正常的S形窄磁滞回线,饱和磁化强度为52.48 emu·g−1。曲线的剩磁和矫顽力几乎为零,表明MF-SBC具有超顺磁特性。MF-SBC的超顺磁行为有利于同步吸附水溶液中的磷酸盐和氨氮,并且外部磁场可以从处理溶液中将材料分离出来。如图1(b)所示,可使用磁铁能够将吸附后的MF-SBC从溶液中分离出来,这便于材料的回收。
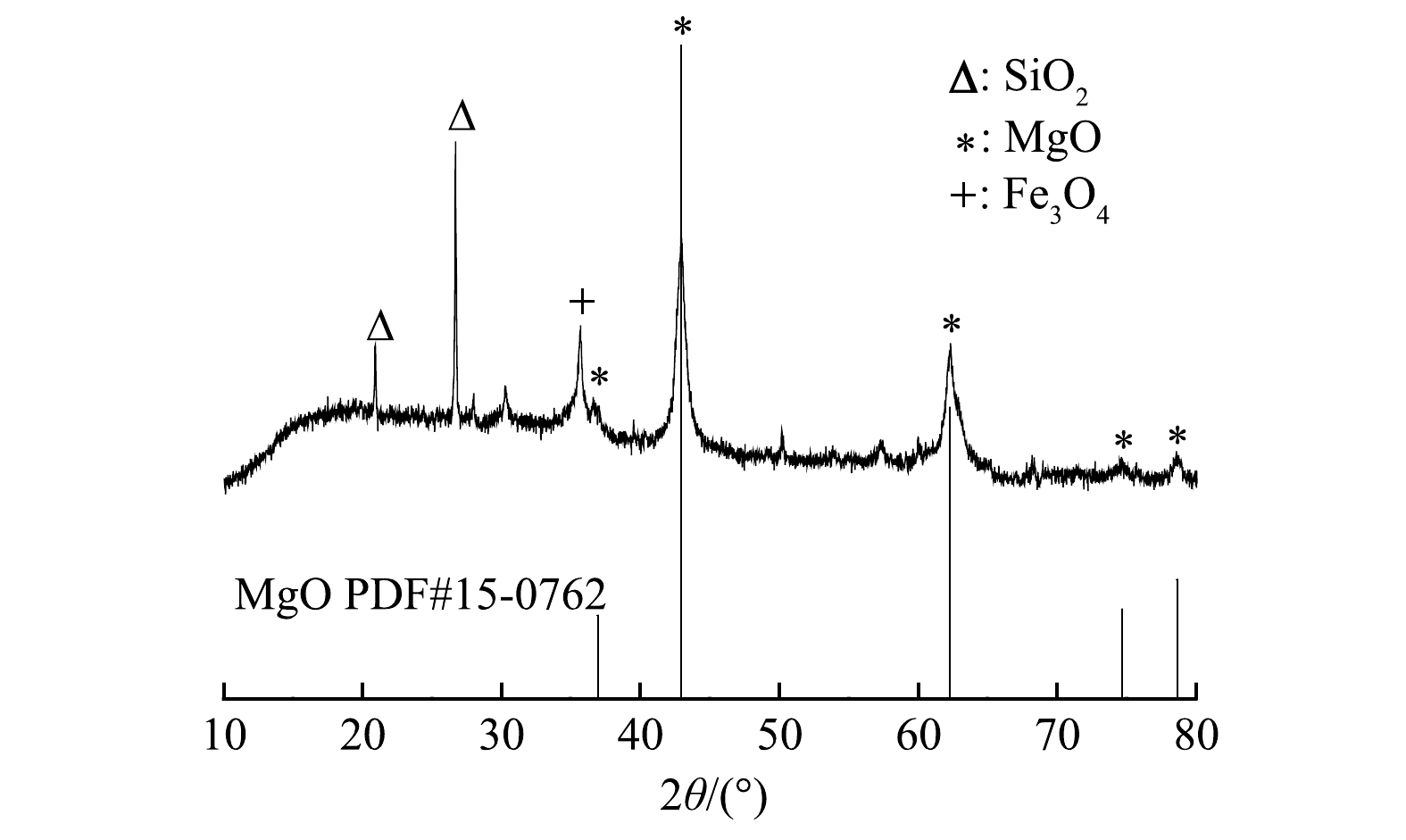
由图2可以看出,在2θ=35.66°处有一个对应于Fe3O4的衍射峰。这表明MF-SBC中含有Fe3O4,解释了MF-SBC具有磁性的原因。在2θ为20.98°和26.66°处可以观察到SiO2的衍射峰,这是污泥中自带的杂质成分。此外,在2θ为42.94°、62.30°、74.67°和78.64°处均有衍射峰,与MgO标准卡片 (PDF#45-0946) 高度吻合,可判定生物炭中含有MgO,表明MF-SBC是富含MgO颗粒的磁性污泥生物炭。
吸附剂的表面形貌和微观结构特征与材料的吸附性能有着密切联系,是材料表征的一个重要参数。图3为MF-SBC在不同放大倍数下的SEM表征结果。可见,随着放大倍数的增加,MF-SBC表面呈不规则形态,具有稠密生长的团聚颗粒。这可能是嵌入到生物炭表面的MgO颗粒,致使MF-SBC比表面积大,反应位点多,镁离子可以在溶液中不断释放,因此有利于对氨氮和磷酸盐同步回收。
MF-SBC吸附氨氮和磷酸盐前后的N2吸附脱附曲线见图4。由图4可以看出,吸附前后MF-SBC对氮气的吸附脱附属于IV型吸附等温线,在相对压力为0.4~1.0时有明显的滞回环,且吸附后的滞回环大于吸附前。这表明吸附后MF-SBC上部分介孔变成了微孔[20-21]。此外,从孔径分布图看出,吸附后MF-SBC表面孔径有所变小。
利用BET法可以计算同步吸附氨氮和磷酸盐前后MF-SBC的总孔体积、平均孔径和比表面积。可以看出,MF-SBC的总孔体积、平均孔径和比表面积分别为0.16 m3·g−1、10.99 nm和58.99 m2·g−1,平均孔径为2~50 nm,属于介孔结构。在MF-SBC同步吸附氨氮和磷酸盐后,总孔体积、平均孔径和比表面积均有所下降,分别为0.12 m3·g−1、10.24 nm和48.69 m2·g−1,与N2吸附脱附曲线的分析相一致,表明同步吸附氨氮和磷酸盐后,MF-SBC表面孔隙发生堵塞,这可能由于MF-SBC将磷酸盐和氨氮吸附到其表面并占据了部分吸附位点。
-
如图5(a)所示,当pH为3.0~11.0,MF-SBC对氨氮的吸附量大于对磷酸盐的吸附量,氨氮和磷酸盐的吸附量都是随着pH的升高而变大。在pH为9.0~11.0时,氨氮和磷酸盐的吸附量随着pH的升高而降低,在pH为9时,氨氮和磷酸盐最大吸附量分别为85.8 mg·g−1和209.95 mg·g−1。△pH为吸附后溶液的pH减去溶液初始pH。如图5(b)所示,在初始溶液pH为8.8时,△pH为0,表明MF-SBC的pHpzc在8.8左右。另外,MF-SBC的Zeta电位值变化也证实了这一点。因此,当溶液pH小于8.8时,MF-SBC表面带正电荷,且pH越低,MF-SBC所带的正电荷越强越易吸引带有负电荷的磷酸盐而排斥带有正电荷的铵根离子。反之,当溶液pH大于8.8时,MF-SBC表面则呈负电荷。在溶液pH为11时,氨氮表现出极低的吸附量,这可能是高pH溶液会使大量的NH4+水解并挥发成NH3,进而降低氨氮吸附量。
-
由图6可以看出,MF-SBC的投加量从0.1 g·L−1加至0.3 g·L−1时,氨氮的吸附量有明显的增加,而继续增加投加量后,氨氮的吸附量开始减小。对于磷酸盐的吸附,从0.1 g·L−1加至0.3 g·L−1时,磷酸盐的吸附量缓慢减小,几乎无变化;继续增加投加量后,磷酸盐吸附量有大幅度的减小。因此,当MF-SBC的投加量为0.3 g·L−1时,同步吸附溶液中氨氮和磷酸盐的效果最佳。造成这种现象的原因可能是,MF-SBC投加量的增大降低了磷酸盐和氨氮与单位质量MF-SBC比值,此时,吸附位点仍然保持不饱和状态[22]。
-
图7反映了在初始pH为9的情况下,接触时间对MF-SBC同步吸附溶液中氮磷的影响。可见,在0~60 min内,MF-SBC对氨氮和磷酸盐的吸附量急剧上升,再缓慢增加到120 min后达到吸附平衡状态,此时氨氮和磷酸盐的吸附量分别达到了103.12 mg·g−1和205.07 mg·g−1。在MF-SBC同步吸附溶液中氨氮和磷酸盐的初期,MF-SBC表面含有大量的吸附位点和镁离子,反应容易进行,但随着反应的进行,MF-SBC表面吸附位点被逐渐占据,且镁离子浓度也随之减低,不利于反应的进行。
为进一步分析MF-SBC对氨氮和磷酸盐的吸附过程,采用准一级动力学和准二级动力学模型对吸附数据进行拟合,结果见图8和表2。对于氨氮和磷酸盐,准二级动力学模型的拟合系数均高于准一级动力学模型,Qe的计算值与实际值更相近,表明吸附过程更符合准二级动力学,这说明吸附过程是以化学吸附为主,其中可能包括阳离子交换、络合和沉淀[23]。另外我们还拟合了吸附数据的颗粒内扩散模型(图8(c)和表3),其中S1阶段表示离子的表面扩散阶段,S2表示离子的颗粒内扩散阶段,S3表示离子的吸附平衡阶段[24]。结果表明,S1阶段所占时间最长,因此吸附过程是以S1阶段的表面扩散为主。此外,S1阶段的拟合直线斜率不为0,这表明吸附过程不仅仅只有表面扩散阶段,还包含颗粒内扩散阶段[21,25]。
-
在实际废水中往往存在很多共存离子,由于这些离子由于本身都带有电荷,可能会与水中的氨氮和磷酸盐形成竞争吸附关系。Ca2+、Na+、SO42− 3种离子对MF-SBC同步吸附氨氮和磷酸盐的影响,结果见图9。由图9可以看出,Ca2+对MF-SBC吸附磷酸盐基本无影响,对氨氮的吸附有抑制作用,且Ca2+的浓度越高,抑制效果越明显,当Ca2+的浓度为100 mg·L−1时,MF-SBC对氨氮的吸附量仅为56.33 mg·g−1。这可能是由于Ca2+会与磷酸盐发生反应生成羟基磷灰石(Ca(PO4)6(OH)2)[25],不利于鸟粪石沉淀的生成。此外,Ca2+在溶液中易水解释放H+,降低溶液pH,从而抑制氨氮的去除。Na+、SO42−对MF-SBC吸附氨氮和磷酸盐几乎没有影响。
-
MF-SBC的回收率以及用于循环实验后对离子的去除率是影响材料后续应用成本的重要指标。MF-SBC经过Fe改性,4次循环实验后,回收率依然高达62.99%(图10)。结果表明,在循环实验中,MF-SBC对氨氮的吸附容量由103.11 mg·g−1降至75.63 mg·g−1,对磷酸盐的吸附容量由204.97 mg·g−1降至173.22 mg·g−1。这可能是由于在去除氨氮和磷酸盐的过程中Mg2+的消耗所导致的,但总的来说,MF-SBC对氨氮和磷酸盐吸附容量分别仅下降了26.65%和15.49%,仍具有较高的去除率。
-
在溶液初始pH为9时,磁性污泥生物炭MF-SBC对氨氮和磷酸盐的吸附机理主要有以下几种。表面吸附机理:MF-SBC作为负载MgO颗粒的污泥基生物炭材料自身具有较大的比表面积和孔体积,能够将氨氮和磷酸盐吸附到其表面。离子交换机理:MF-SBC材料中除Mg2+外,还包含一些来自污泥的碱金属,如Na+、Ca2+和K+等。这些金属离子附着在生物炭表面会与溶液中的NH4+发生离子交换反应,如反应式(5)和式(6)所示。此外,MF-SBC中的羟基也会与磷酸盐离子发生离子交换反应,如反应式(7)所示。为了判断离子交换去除氮磷的可能性,对MF-SBC在pH为9的条件下进行了浸渍(图11)。结果表明,只有微量Ca2+存在溶液中,有部分氨氮是通过离子交换固定在MF-SBC表面。但Ca2+数量极少,以Ca2+为主的离子交换并不是主要的去除机理。此外,由Mg1s的XPS图谱中可以看到,吸附后Mg1s的峰面积相比于吸附前降低了26.14%,这也是在吸附过程中Mg以离子交换形式去除氨氮和磷酸盐的直接证据[26-28]。
鸟粪石沉淀机理。在初始pH=9的条件下,表面吸附和离子交换对氨氮和磷酸盐的回收能力有限,并不能做到生物炭对氨氮和磷酸盐的高效同步回收,其主要回收方式是鸟粪石沉淀法。当初始溶液pH=9时,磷酸盐的主要存在形态为HPO42−和PO43−,氨氮在溶液中的存在形态是NH4+。此时,当MF-SBC中的镁离子释放到溶液中,会与磷酸盐离子和NH4+反应形成鸟粪石沉淀(式(8)和式(9))。可见,在形成鸟粪石过程中会有一定量的H+释放到溶液中,解释了反应后溶液pH变低的现象,同时H+的增加,也促进了MF-SBC表面MgO的水解,进一步促进了鸟粪石沉淀反应的发生。
通过红外和XRD分析可以鸟粪石沉淀的生成。由图12(a)可以看出,对于吸附前后的MF-SBC,在3 458 cm−1的宽吸收带和1 634 cm−1的峰归因于H—O—H水分子的伸缩峰与弯曲振动峰。吸附后的谱图在1 438 cm−1和1 079 cm−1处生成了2个明显的特征吸收峰,分别对应于NH4+和PO43−,这表明NH4+和PO43−都存在于回收的MF-SBC中。图12(b)是回收产物的XRD谱图。可以看出在2θ为14.98°、15.92°、20.87°、30.24°、31.90°、33.17°和46.36°处有明显的高强度衍射峰,与鸟粪石的标准卡片 (PDF#15-0762) 基本吻合。表明回收产物中含有大量的鸟粪石晶体,进一步证实了鸟粪石沉淀是MF-SBC同步吸附溶液中氮磷的主要作用机制。
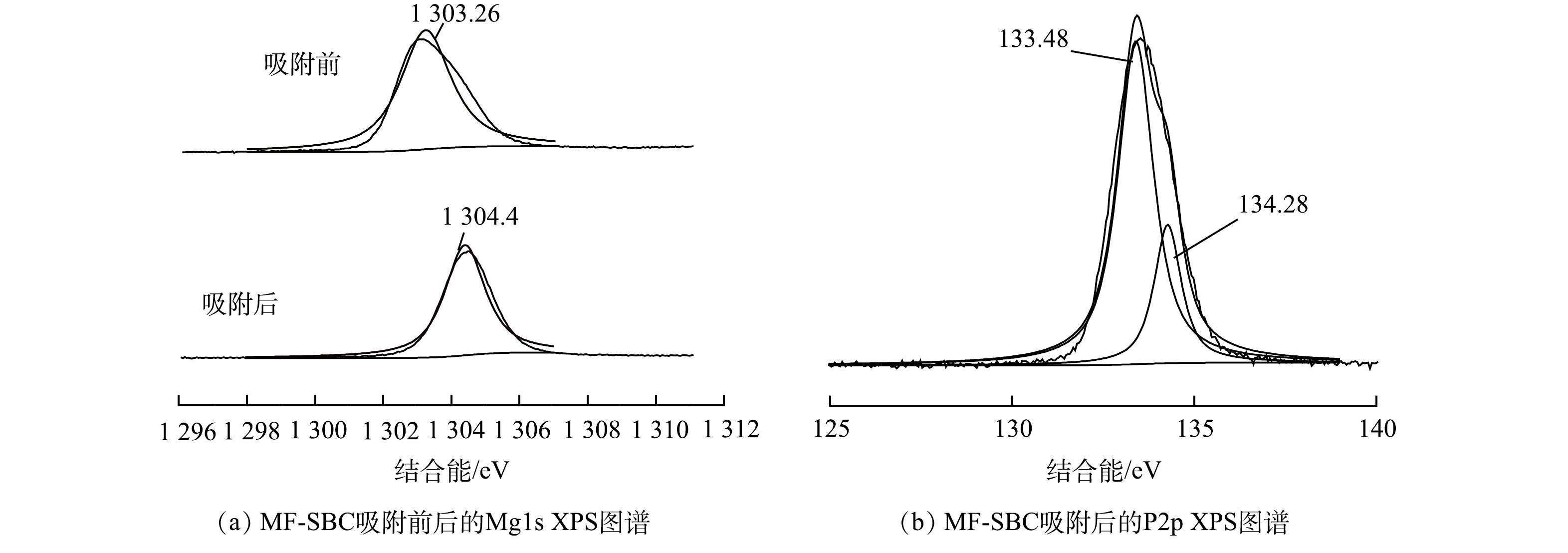
由图13(a)可以看出,吸附前后MF-SBC中Mg元素的特征峰分别在1 303.26 eV和1 304.42 eV处,归属于不同的Mg形态。吸附前位于1 303.26 eV处的特征峰表明Mg以金属氧键的方式,即氧化镁的形式存在;而反应之后在1 304.42 eV处的特征峰则归属于Mg的非金属形态,表明镁离子参与反应生成了鸟粪石沉淀。由图13(b)可见,不同形态的磷所处的结合能不同,吸附后MF-SBC的P2p峰可分为133.48 eV和134.28 eV 2个单峰,分别是鸟粪石沉淀表面的P=O和P—OH官能团。
-
1)通过煅烧法,成功合成了磁性污泥基生物炭MF-SBC,其表面含有大量的氧化镁颗粒,比表面积为58.99 m2·g−1,饱和磁化强度为52.48 emu·g−1。
2)在MF-SBC投加量为0.3 g·L−1,溶液初始pH为9时,MF-SBC同步吸附氨氮和磷酸盐的吸附量最大,分别为103.12 mg·g−1和205.07 mg·g−1,且吸附过程符合准二级动力学方程。Ca2+、Na+、SO42− 3种离子对磷酸盐的吸附无影响,而Ca2+和SO42−对氨氮的吸附有抑制作用。
3)在初始溶液pH=9的条件下,MF-SBC对水溶液中氨氮和磷酸盐同步吸附的作用机制包括表面吸附、离子交换和鸟粪石沉淀,其中鸟粪石沉淀为主要作用。
磁性污泥基生物炭的制备及其对水溶液中氮磷的同步回收
Preparation of magnetic sludge-based biochar for the simultaneous recovery of nitrogen and phosphorus from aqueous solution
-
摘要: 以城市污泥为原料与MgCl2和FeSO4复合,并热解碳化合成磁性污泥基生物炭(MF-SBC),用于水中氮磷的同步回收研究,分别考察了MF-SBC投加量、初始pH、接触时间和共存离子对氮磷回收性能的影响,同时通过SEM、XRD、BET、XPS和FTIR表征了MF-SBC的组成、形貌和官能团等,并对反应过程进行了动力学拟合。结果表明,当MF-SBC投加量为0.3 g·L−1、溶液初始pH为7、反应时间为720 min时,MF-SBC对水溶液中氨氮和磷酸盐的回收效果最佳,吸附量分别为103.12 mg·g−1和205.07 mg·g−1,并且MF-SBC对水中氨氮和磷酸盐的回收过程均符合准二级动力学模型。Ca2+、Na+、SO42对MF-SBC回收磷酸盐几乎没有影响,Ca2+和SO42-对氨氮的回收有抑制作用。MF-SBC对氮磷的回收机制包括表面吸附、离子交换和鸟粪石沉淀,其中以鸟粪石沉淀为主。Abstract: In this study, municipal sludge was taken as raw material combined with MgCl2 and FeSO4, magnetic sludge-based biochar (MF-SBC) was synthesized by pyrolysis carbonization, which was used to simultaneously recover nitrogen and phosphorus in water. Meanwhile, the effects of dosage, initial pH, contact time and coexisting ions on the recovery performance were investigated. The composition, morphology and functional groups of MF-SBC before and after adsorption were characterized by SEM, XRD, BET, XPS and FTIR, and the reaction process was fitted by kinetic models. The results showed that MF-SBC had the best recovery effect of ammonia nitrogen and phosphate in aqueous solution, and the maximum adsorption capacities were 103.12 mg·g−1 and 205.07 mg·g−1, respectively, when MF-SBC dosage was 0.3 g·L−1, the initial pH was 7, and the reaction time was 720 min, and the recovery process of ammonia nitrogen and phosphate in water by MF-SBC conformed to the pseudo-second-order kinetic model. Ca2+, Na+ and SO42- had slight effect on phosphate recovery by MF-SBC, Ca2+ and SO42− had an inhibitory effect on ammonia recovery. The recovery mechanism included surface adsorption, ion exchange and struvite precipitation, which was dominated by struvite precipitation.
-
Key words:
- sludge-based biochar /
- nitrogen and phosphorus /
- simultaneous recovery /
- struvite
-
氮、磷污染是造成水体富营养化的主要原因,同时氮和磷也是一种重要资源,是生产农肥和畜禽饲料的原料。全球磷资源十分有限,且单向流动、难以再生。在现有技术、经济水平条件下,全球探明可供开采的磷矿资源不足100 a[1]。富含氮磷元素废水的排放不仅会造成氮磷的流失,还会造成地表水体富营养化。因此,高效回收废水中的氮磷资源,已成为相关领域的研究热点之一。
目前的氮磷共存水体主要是养殖废水、食品废水和市政污水。其中山东某食品公司产生的废水氨氮和总磷含量高达160 mg·L−1和100 mg·L−1,经过初沉池、UASB池、A/O池和二沉池处理后氨氮含量显著降低,但总磷含量依然高达77 mg·L−1[2]。此外,在甘肃某养牛场养殖废水中氨氮和磷酸盐质量浓度也高达1 304 mg·L−1和146 mg·L−1[3]。目前,最常用的脱氮除磷工艺为氧化沟工艺、A2O工艺和SBR工艺。但上述工艺对污水水质的稳定性要求较高,水质的大幅度变化会影响微生物脱氮除磷效果。此外,污水经二级处理后增加深度处理方能较好净化水质,但随之而来的是高昂的化学添加剂成本以及基建和电力费用。因此,寻找一种廉价的清洁材料用于同步、高效去除氮磷对于治理氮磷废水至关重要。
目前,磷酸铵镁(magnesium ammonium phosphate,MAP)结晶法往往具备同步实现对氮、磷的高效脱除和有效回收,广泛用于尿液、垃圾渗滤液、焦化废水的处理[4-6]。MAP英文俗名为struvite,中文俗称鸟粪石,化学成分为MgNH4PO4·6H2O,是一种较难溶于水的无色、白色(脱水后)、黄色、棕色或浅灰色的晶体,且含有氮、磷植物营养元素,是一种很好的缓释化肥[7-8]。其基本原理是在沼液中投加Mg2+,在碱性条件下使沼液中的PO43−、HPO42−、H2PO4−及NH4+与Mg2+反应成生MgNH4PO4·6H2O(struvite,即鸟粪石),从而回收水体的氮磷[9]。传统的MAP结晶法回收率较低,如磷结晶率达90%以上时,氨氮结晶率仅为13%左右,难以达到两者同步高效结晶[10]。鸟粪石的形成需要在较高的溶液pH条件下,需投加含Mg2+沉淀剂和碱溶液,原材料成本过高限制该方法的使用,且反应过后的材料回收困难[11]。HUANG等[12]利用镁盐改性天然沸石同步去除废水中的氮磷获得良好的去除效果。针对以上问题,选择一种自身碱性高、成本低和易回收的材料尤为重要。
常见镁来源有Mg(OH)2、MgSO4、MgCl2等。其中,MgO由于其安全稳定且自身碱性高等优点,受到广泛应用[10, 12-13]。生物炭比表面积大、离子交换能力强,可以去除水中的污染物[13-14]。并且生物炭表面呈负电荷,可以吸附水中的氨氮,但通常不能吸附磷酸盐。利用金属氧化物进行生物炭进行改性,可以有效改善其对磷的吸附性能[14]。生物质原料包括农业和森林残余物及其副产品(水稻秸秆[15]、小麦秸秆[16]、玉米芯[17]和菜叶[18]等)、动物粪便、造纸厂废料、城市固体废物和污泥[19-20]。选择市政污泥作为原料制备污泥基生物炭,具有来源稳定、成本低等优点,同时也为污泥的资源化利用提供了一种途径,具有良好社会意义。
因此,本研究开发了一种以镁离子为靶向供给、碱性调控能力强、成本低廉且易于回收的新型磁性污泥基生物炭复合材料(Mg/Fe sludge biochar, MF-SBC),用于废水中氮磷的同步回收。考察了初始pH、接触时间、共存离子、投加量对氮磷回收效果的影响,采用动力学模型等分析了回收特征,并结合XRD、XPS、SEM等多种表征手段探讨了新型磁性污泥基生物炭对水溶液中氮磷同步回收的作用机制,为其实际应用提供参考。
1. 材料与方法
1.1 实验材料
实验所用试剂包括七水合硫酸亚铁(FeSO4·7H2O)、六水合氯化镁(MgCl2·6H2O)、氢氧化钠(NaOH)、氯化铵(NH4Cl)和磷酸二氢钾(KH2PO4),以上试剂均为化学纯或分析纯。使用去离子水制备所有溶液。本实验原料取自中国江苏省武进区武南污水厂的脱水污泥,将脱水污泥在105 ℃条件下烘至恒重,研磨过200目筛,置于干燥器中备用。通过X射线荧光光谱仪检测到污泥粉末中主要的无机化学成分有SiO2、Al2O3、CaO、MgO、Fe2O3和P2O5。
1.2 磁性污泥基生物炭的制备
MF-SBC的制备具体步骤如下:准确称取2.5 g干燥的污泥粉末加入到50 mL 0.2 mmol·L−1 FeSO4·7H2O溶液中浸泡并振荡6 h;将污泥/FeSO4混合溶液放入烘箱中经80 ℃烘干并研磨过200目筛,之后将其加入50 mL 1.25 mmol·L−1 MgCl2·6H2O溶液,并进行磁力搅拌30 min;随后,向上一步得到的混合溶液中缓慢加入50 mL 2.5 mmol·L−1 NaOH溶液并磁力搅拌12 h,紧接着陈化24 h;过滤以上溶液后所得沉淀物用去离子水反复清洗至中性,再经80 ℃烘干;将得到的干燥沉淀物放置管式炉中以5 ℃·min−1慢速升温至450 ℃,并在该温度下持续煅烧2 h,得到磁性MF-SBC。
1.3 序批式实验
分别使用KH2PO4和NH4Cl配置磷酸盐和氨氮储备溶液,并稀释至实验所需浓度。对于每批次实验,将一定量的MF-SBC材料加入到100 mL含有磷酸盐和氨氮的锥形瓶中,并用0.1 mmol·L−1 HCl和NaOH调节溶液至所需pH,之后将密封的锥形瓶置于恒温振荡器中振荡。反应结束后,取上层清液,用0.45 μm滤膜过滤后用UV-vis分光光度计(pharo300,merck, 德国)检测。分别考察了初始pH、氨氮和磷酸盐的质量浓度、MF-SBC投加量和反应时间对MF-SBC同步吸附氨氮和磷酸盐的影响,各批次实验的详细实验条件见表1。
表 1 MF-SBC同步吸附氨氮和磷酸盐的详细实验条件Table 1. Experimental conditions for the simultaneous adsorption of nitrogen and phosphate by MF-SBC影响因素 时间/min 氨氮质量浓度/(mg·L−1) P质量浓度/(mg·L−1) pH 共存离子 MF-SBC质量浓度/(g·L−1) pH 720 160 80 3~11 — 0.3 MF-SBC投加量 720 160 80 9 — 0.1~0.5 共存离子 720 160 80 9 Ca2+、Na+、SO42− 0.3 反应时间 1~720 160 80 9 — 0.3 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 分析方法
1)生物炭表征。采用阶级开门扫描方式进行分析,扫描角2θ范围为10°~80°,工作电压和电流分别为60 kV和300 mA;分别利用扫描电子显微镜(SEM,SUPRA5,德国)和快速全自动比表面和孔径分析仪(BET,Autosorb-iQ2-MP,美国)来观察和测定微观形貌和比表面积;使用X射线光电子能谱仪(XPS,EscaLab 250Xi,美国)和能量色散X射线光谱(EDX)分析样品结构表面处的局部元素组成。在400~4 000 cm−1内通过傅里叶光谱仪(FTIR,Nicolet IS5 美国)记录红外光谱,鉴定所制备样品官能团。
2)溶液中污染物浓度的测定。采用国标钼酸铵分光光度法(GB11893-89)和纳氏试剂分光光度法(GB11893-89)测定溶液中磷和氨氮含量,每组3个平行样,到达反应平衡时,利用式(1)计算MF-SBC的去除量。
Qe=VM(C0−Ce) (1) 式中:Qe为吸附剂的吸附量,mg·g−1;C0为溶液的初始质量浓度,g·L−1;Ce为吸附后的溶液质量浓度,mg·L−1;V为溶液的体积,L;M为吸附剂质量,g。
1.5 动力学模型
为分析MF-SBC对磷酸盐和氨氮的吸附动力学情况,采用准一级动力学方程(式(2))、准二级动力学方程(式(3))和颗粒内扩散方程(式(4))对其进行拟合。
ln(Qe−Qt)=lnQe−k1t (2) 1Qe−Qt=1Qe+k2t (3) Qt=kpt0.5+C (4) 式中:t表示吸附时间,min;Qt表示t时吸附剂的吸附量,mg·g−1;Qe表示吸附平衡的吸附量,mg·g−1;k1为一级速率常数,min−1;k2为二级速率常数,g·(mg·min)−1;kp表示颗粒内扩散速率常数,mg·(mg·min)−1;C是常数,为颗粒内扩散方程的截距。
1.6 MF-SBC循环实验
吸附实验结束后,过滤收集MF-SBC,用1 mol·L−1 NaOH溶液超声30 min并静置浸渍1 h,再用去离子水冲洗至pH为中性后烘干,干燥保存并用于下一次的循环使用。
2. 结果与讨论
2.1 生物炭的表征
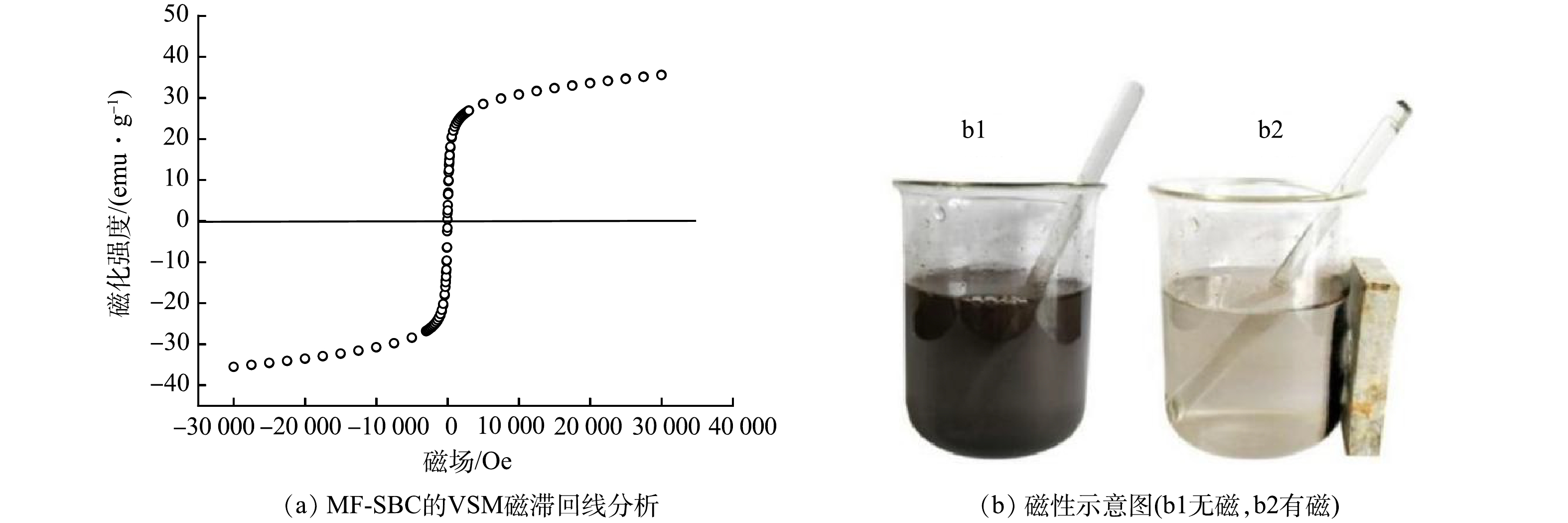
为了确定MF-SBC的磁性,在室温下通过振动样品磁强计(VSM)来测量材料的磁性,得到磁滞回线(图1(a))。由图1(a)可以看出,MF-SBC的曲线显示出正常的S形窄磁滞回线,饱和磁化强度为52.48 emu·g−1。曲线的剩磁和矫顽力几乎为零,表明MF-SBC具有超顺磁特性。MF-SBC的超顺磁行为有利于同步吸附水溶液中的磷酸盐和氨氮,并且外部磁场可以从处理溶液中将材料分离出来。如图1(b)所示,可使用磁铁能够将吸附后的MF-SBC从溶液中分离出来,这便于材料的回收。
由图2可以看出,在2θ=35.66°处有一个对应于Fe3O4的衍射峰。这表明MF-SBC中含有Fe3O4,解释了MF-SBC具有磁性的原因。在2θ为20.98°和26.66°处可以观察到SiO2的衍射峰,这是污泥中自带的杂质成分。此外,在2θ为42.94°、62.30°、74.67°和78.64°处均有衍射峰,与MgO标准卡片 (PDF#45-0946) 高度吻合,可判定生物炭中含有MgO,表明MF-SBC是富含MgO颗粒的磁性污泥生物炭。
吸附剂的表面形貌和微观结构特征与材料的吸附性能有着密切联系,是材料表征的一个重要参数。图3为MF-SBC在不同放大倍数下的SEM表征结果。可见,随着放大倍数的增加,MF-SBC表面呈不规则形态,具有稠密生长的团聚颗粒。这可能是嵌入到生物炭表面的MgO颗粒,致使MF-SBC比表面积大,反应位点多,镁离子可以在溶液中不断释放,因此有利于对氨氮和磷酸盐同步回收。
MF-SBC吸附氨氮和磷酸盐前后的N2吸附脱附曲线见图4。由图4可以看出,吸附前后MF-SBC对氮气的吸附脱附属于IV型吸附等温线,在相对压力为0.4~1.0时有明显的滞回环,且吸附后的滞回环大于吸附前。这表明吸附后MF-SBC上部分介孔变成了微孔[20-21]。此外,从孔径分布图看出,吸附后MF-SBC表面孔径有所变小。
 图 4 吸附前后MF-SBC脱吸附曲线和孔径变化Figure 4. N2 adsorption and desorption curves and change in pore diameter of MF-SBC before and after adsorption
图 4 吸附前后MF-SBC脱吸附曲线和孔径变化Figure 4. N2 adsorption and desorption curves and change in pore diameter of MF-SBC before and after adsorption利用BET法可以计算同步吸附氨氮和磷酸盐前后MF-SBC的总孔体积、平均孔径和比表面积。可以看出,MF-SBC的总孔体积、平均孔径和比表面积分别为0.16 m3·g−1、10.99 nm和58.99 m2·g−1,平均孔径为2~50 nm,属于介孔结构。在MF-SBC同步吸附氨氮和磷酸盐后,总孔体积、平均孔径和比表面积均有所下降,分别为0.12 m3·g−1、10.24 nm和48.69 m2·g−1,与N2吸附脱附曲线的分析相一致,表明同步吸附氨氮和磷酸盐后,MF-SBC表面孔隙发生堵塞,这可能由于MF-SBC将磷酸盐和氨氮吸附到其表面并占据了部分吸附位点。
2.2 初始pH对吸附性能的影响
如图5(a)所示,当pH为3.0~11.0,MF-SBC对氨氮的吸附量大于对磷酸盐的吸附量,氨氮和磷酸盐的吸附量都是随着pH的升高而变大。在pH为9.0~11.0时,氨氮和磷酸盐的吸附量随着pH的升高而降低,在pH为9时,氨氮和磷酸盐最大吸附量分别为85.8 mg·g−1和209.95 mg·g−1。△pH为吸附后溶液的pH减去溶液初始pH。如图5(b)所示,在初始溶液pH为8.8时,△pH为0,表明MF-SBC的pHpzc在8.8左右。另外,MF-SBC的Zeta电位值变化也证实了这一点。因此,当溶液pH小于8.8时,MF-SBC表面带正电荷,且pH越低,MF-SBC所带的正电荷越强越易吸引带有负电荷的磷酸盐而排斥带有正电荷的铵根离子。反之,当溶液pH大于8.8时,MF-SBC表面则呈负电荷。在溶液pH为11时,氨氮表现出极低的吸附量,这可能是高pH溶液会使大量的NH4+水解并挥发成NH3,进而降低氨氮吸附量。
 图 5 溶液初始pH对MF-SBC同步吸附氮磷的影响和MF-SBC的Zeta电位Figure 5. Effect of initial pH on the adsorption capacity and zeta potential of MF-SBC
图 5 溶液初始pH对MF-SBC同步吸附氮磷的影响和MF-SBC的Zeta电位Figure 5. Effect of initial pH on the adsorption capacity and zeta potential of MF-SBC2.3 MF-SBC投加量对吸附性能的影响
由图6可以看出,MF-SBC的投加量从0.1 g·L−1加至0.3 g·L−1时,氨氮的吸附量有明显的增加,而继续增加投加量后,氨氮的吸附量开始减小。对于磷酸盐的吸附,从0.1 g·L−1加至0.3 g·L−1时,磷酸盐的吸附量缓慢减小,几乎无变化;继续增加投加量后,磷酸盐吸附量有大幅度的减小。因此,当MF-SBC的投加量为0.3 g·L−1时,同步吸附溶液中氨氮和磷酸盐的效果最佳。造成这种现象的原因可能是,MF-SBC投加量的增大降低了磷酸盐和氨氮与单位质量MF-SBC比值,此时,吸附位点仍然保持不饱和状态[22]。
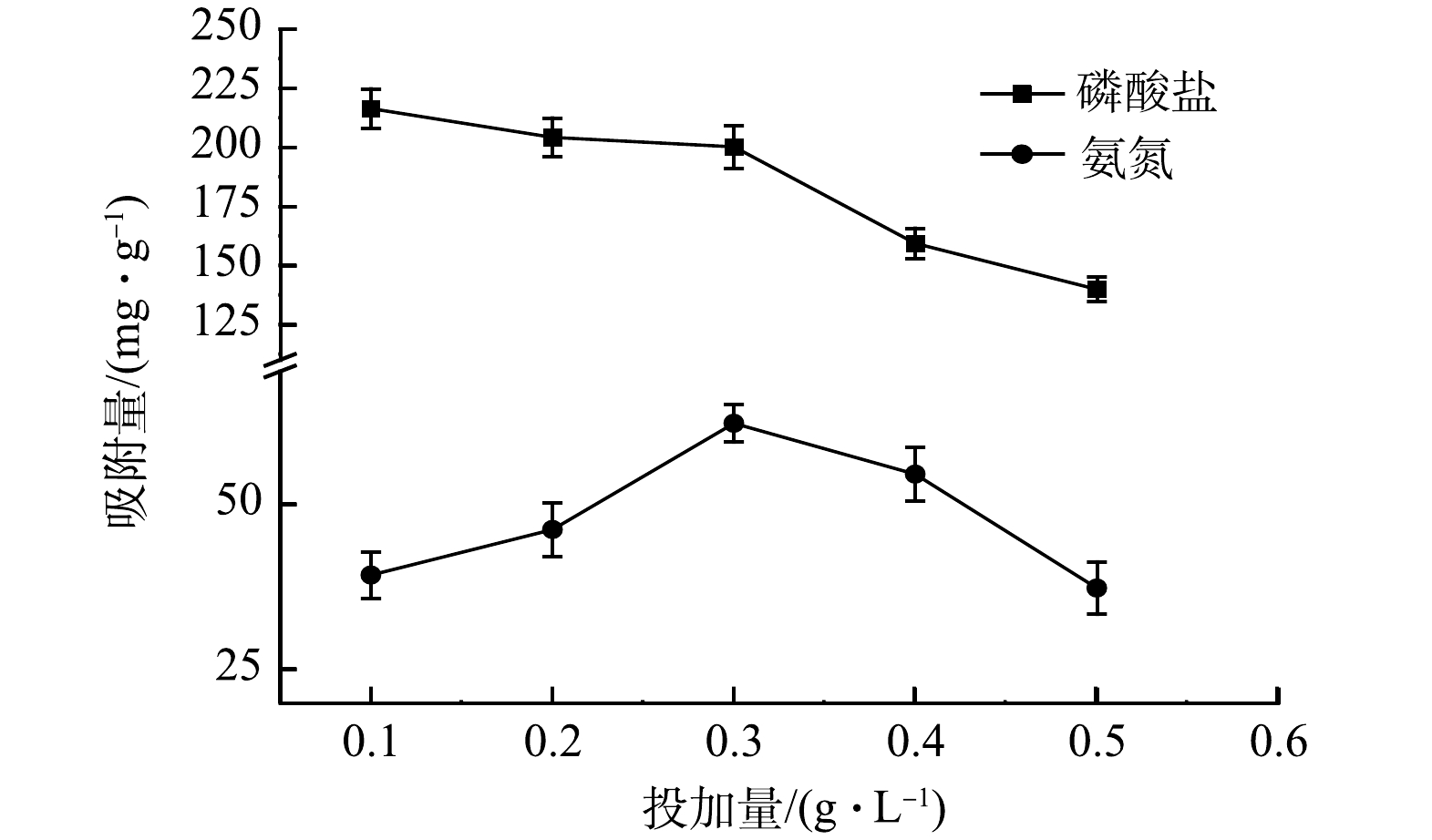 图 6 投加量对MF-SBC同步吸附氮磷的影响Figure 6. Effect of dosage on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 6 投加量对MF-SBC同步吸附氮磷的影响Figure 6. Effect of dosage on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC2.4 反应时间对吸附性能的影响及吸附动力学
图7反映了在初始pH为9的情况下,接触时间对MF-SBC同步吸附溶液中氮磷的影响。可见,在0~60 min内,MF-SBC对氨氮和磷酸盐的吸附量急剧上升,再缓慢增加到120 min后达到吸附平衡状态,此时氨氮和磷酸盐的吸附量分别达到了103.12 mg·g−1和205.07 mg·g−1。在MF-SBC同步吸附溶液中氨氮和磷酸盐的初期,MF-SBC表面含有大量的吸附位点和镁离子,反应容易进行,但随着反应的进行,MF-SBC表面吸附位点被逐渐占据,且镁离子浓度也随之减低,不利于反应的进行。
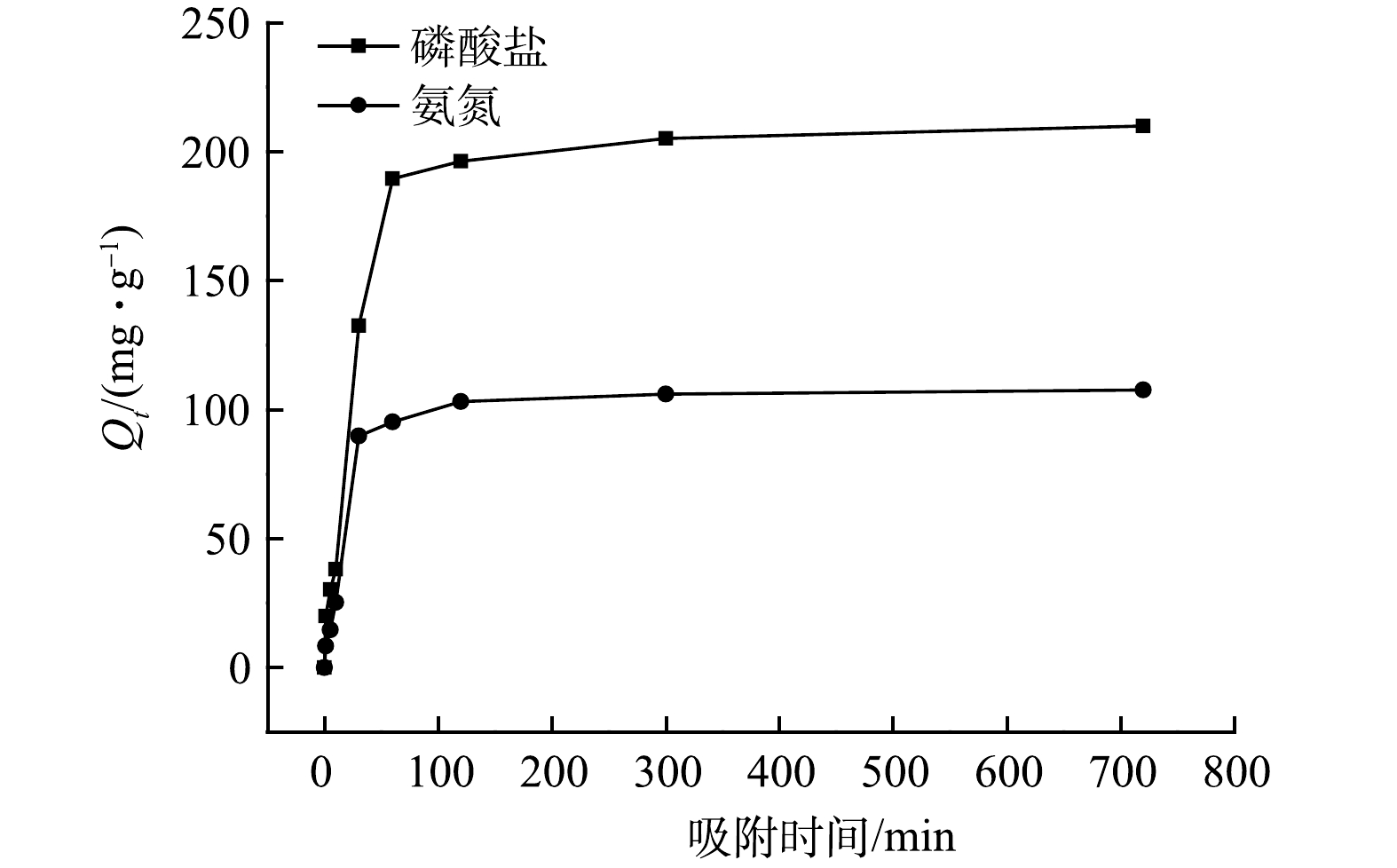 图 7 接触时间对MF-SBC同步吸附氮磷的影响Figure 7. Effect of the contact time on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 7 接触时间对MF-SBC同步吸附氮磷的影响Figure 7. Effect of the contact time on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC为进一步分析MF-SBC对氨氮和磷酸盐的吸附过程,采用准一级动力学和准二级动力学模型对吸附数据进行拟合,结果见图8和表2。对于氨氮和磷酸盐,准二级动力学模型的拟合系数均高于准一级动力学模型,Qe的计算值与实际值更相近,表明吸附过程更符合准二级动力学,这说明吸附过程是以化学吸附为主,其中可能包括阳离子交换、络合和沉淀[23]。另外我们还拟合了吸附数据的颗粒内扩散模型(图8(c)和表3),其中S1阶段表示离子的表面扩散阶段,S2表示离子的颗粒内扩散阶段,S3表示离子的吸附平衡阶段[24]。结果表明,S1阶段所占时间最长,因此吸附过程是以S1阶段的表面扩散为主。此外,S1阶段的拟合直线斜率不为0,这表明吸附过程不仅仅只有表面扩散阶段,还包含颗粒内扩散阶段[21,25]。
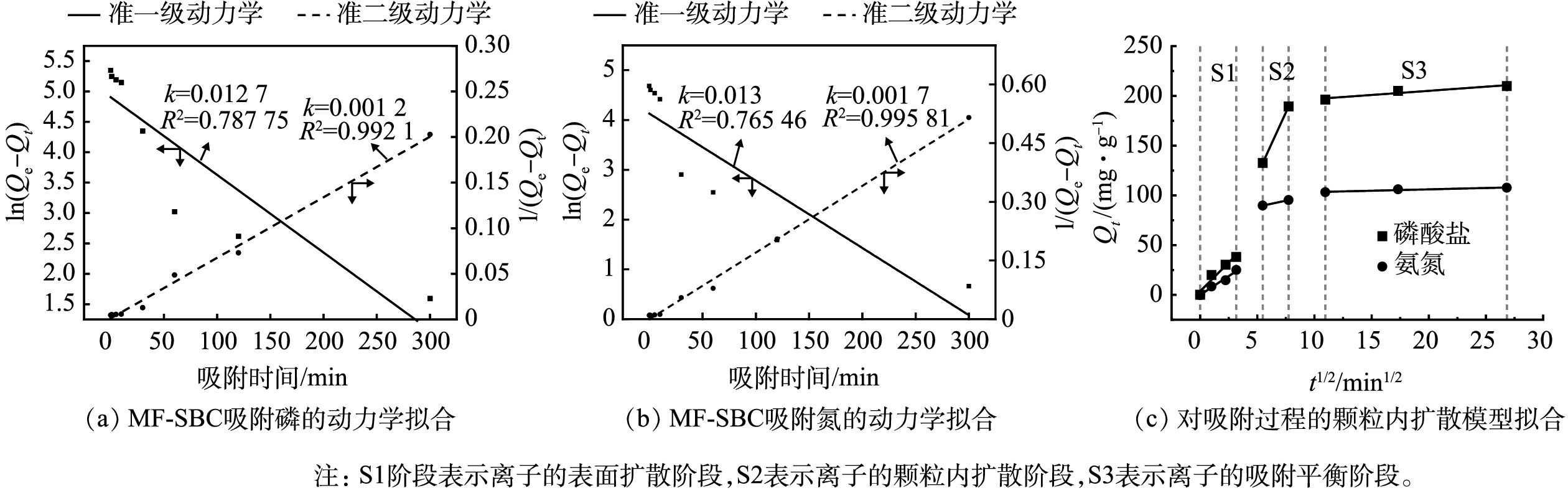 图 8 MF-SBC同步吸附氮磷的动力学拟合Figure 8. Kinetic fitting of the simultaneous adsorption of nitrogen and phosphorus by MF-SBC表 2 MF-SBC同步吸附氮磷的动力学拟合参数Table 2. Kinetic parameters for the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 8 MF-SBC同步吸附氮磷的动力学拟合Figure 8. Kinetic fitting of the simultaneous adsorption of nitrogen and phosphorus by MF-SBC表 2 MF-SBC同步吸附氮磷的动力学拟合参数Table 2. Kinetic parameters for the simultaneous adsorption of nitrogen and phosphorus by MF-SBC污染物 准一级动力学 准二级动力学 Qe /(mg·g−1) k1/ min−1 R2 Qe/(mg·g−1) k1/(g·(mg·min)−1) R2 磷 168.44 0.0127 0.79 203.65 0.0012 0.99 氨氮 83.86 0.013 0.77 101.34 0.0017 0.99 | Show TableDownLoad:
CSV
表 3 MF-SBC同步吸附氮磷的颗粒内扩散模型参数Table 3. Intra particle diffusion model parameters for the simultaneous adsorption of nitrogen and phosphorus by MF-SBC污染物 S1 S2 S3 K R2 K R2 K R2 磷 11.57 0.95 25.07 - 0.83 0.92 氨氮 7.56 0.98 2.43 - 0.28 0.92 | Show TableDownLoad:
CSV
2.5 共存离子对吸附的影响
在实际废水中往往存在很多共存离子,由于这些离子由于本身都带有电荷,可能会与水中的氨氮和磷酸盐形成竞争吸附关系。Ca2+、Na+、SO42− 3种离子对MF-SBC同步吸附氨氮和磷酸盐的影响,结果见图9。由图9可以看出,Ca2+对MF-SBC吸附磷酸盐基本无影响,对氨氮的吸附有抑制作用,且Ca2+的浓度越高,抑制效果越明显,当Ca2+的浓度为100 mg·L−1时,MF-SBC对氨氮的吸附量仅为56.33 mg·g−1。这可能是由于Ca2+会与磷酸盐发生反应生成羟基磷灰石(Ca(PO4)6(OH)2)[25],不利于鸟粪石沉淀的生成。此外,Ca2+在溶液中易水解释放H+,降低溶液pH,从而抑制氨氮的去除。Na+、SO42−对MF-SBC吸附氨氮和磷酸盐几乎没有影响。
 图 9 共存离子对MF-SBC同步吸附氮磷的影响Figure 9. Effects of coexisting ions on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 9 共存离子对MF-SBC同步吸附氮磷的影响Figure 9. Effects of coexisting ions on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC2.6 MF-SBC的回收与循环实验
MF-SBC的回收率以及用于循环实验后对离子的去除率是影响材料后续应用成本的重要指标。MF-SBC经过Fe改性,4次循环实验后,回收率依然高达62.99%(图10)。结果表明,在循环实验中,MF-SBC对氨氮的吸附容量由103.11 mg·g−1降至75.63 mg·g−1,对磷酸盐的吸附容量由204.97 mg·g−1降至173.22 mg·g−1。这可能是由于在去除氨氮和磷酸盐的过程中Mg2+的消耗所导致的,但总的来说,MF-SBC对氨氮和磷酸盐吸附容量分别仅下降了26.65%和15.49%,仍具有较高的去除率。
 图 10 MF-SBC在循环实验中的回收率以及对氨氮和磷酸盐的吸附容量Figure 10. Recovery rate and adsorption capacity to ammonia nitrogen and phosphate by MF-SBC in cycle experiment
图 10 MF-SBC在循环实验中的回收率以及对氨氮和磷酸盐的吸附容量Figure 10. Recovery rate and adsorption capacity to ammonia nitrogen and phosphate by MF-SBC in cycle experiment2.7 吸附机理
在溶液初始pH为9时,磁性污泥生物炭MF-SBC对氨氮和磷酸盐的吸附机理主要有以下几种。表面吸附机理:MF-SBC作为负载MgO颗粒的污泥基生物炭材料自身具有较大的比表面积和孔体积,能够将氨氮和磷酸盐吸附到其表面。离子交换机理:MF-SBC材料中除Mg2+外,还包含一些来自污泥的碱金属,如Na+、Ca2+和K+等。这些金属离子附着在生物炭表面会与溶液中的NH4+发生离子交换反应,如反应式(5)和式(6)所示。此外,MF-SBC中的羟基也会与磷酸盐离子发生离子交换反应,如反应式(7)所示。为了判断离子交换去除氮磷的可能性,对MF-SBC在pH为9的条件下进行了浸渍(图11)。结果表明,只有微量Ca2+存在溶液中,有部分氨氮是通过离子交换固定在MF-SBC表面。但Ca2+数量极少,以Ca2+为主的离子交换并不是主要的去除机理。此外,由Mg1s的XPS图谱中可以看到,吸附后Mg1s的峰面积相比于吸附前降低了26.14%,这也是在吸附过程中Mg以离子交换形式去除氨氮和磷酸盐的直接证据[26-28]。
鸟粪石沉淀机理。在初始pH=9的条件下,表面吸附和离子交换对氨氮和磷酸盐的回收能力有限,并不能做到生物炭对氨氮和磷酸盐的高效同步回收,其主要回收方式是鸟粪石沉淀法。当初始溶液pH=9时,磷酸盐的主要存在形态为HPO42−和PO43−,氨氮在溶液中的存在形态是NH4+。此时,当MF-SBC中的镁离子释放到溶液中,会与磷酸盐离子和NH4+反应形成鸟粪石沉淀(式(8)和式(9))。可见,在形成鸟粪石过程中会有一定量的H+释放到溶液中,解释了反应后溶液pH变低的现象,同时H+的增加,也促进了MF-SBC表面MgO的水解,进一步促进了鸟粪石沉淀反应的发生。
MF−SBC−Mg2++2NH+4→MF−SBC−(NH+4)2+Mg2+ (5) MF−SBC−Ca2++2NH+4→MF−SBC−(NH+4)2+Ca2+ (6) 2(MF−SBC−OH−)+HPO2−4→MF−SBC−HPO2−4+2OH− (7) Mg2++NH+4+HPO2−4→MgNH4PO4⋅6H2O↓+H+ (8) Mg2++NH+4+PO3−4→MgNH4PO4⋅6H2O↓ (9) 通过红外和XRD分析可以鸟粪石沉淀的生成。由图12(a)可以看出,对于吸附前后的MF-SBC,在3 458 cm−1的宽吸收带和1 634 cm−1的峰归因于H—O—H水分子的伸缩峰与弯曲振动峰。吸附后的谱图在1 438 cm−1和1 079 cm−1处生成了2个明显的特征吸收峰,分别对应于NH4+和PO43−,这表明NH4+和PO43−都存在于回收的MF-SBC中。图12(b)是回收产物的XRD谱图。可以看出在2θ为14.98°、15.92°、20.87°、30.24°、31.90°、33.17°和46.36°处有明显的高强度衍射峰,与鸟粪石的标准卡片 (PDF#15-0762) 基本吻合。表明回收产物中含有大量的鸟粪石晶体,进一步证实了鸟粪石沉淀是MF-SBC同步吸附溶液中氮磷的主要作用机制。
 图 12 MF-SBC吸附前后的FT-IR和吸附后的XRD图谱Figure 12. FT-IR spectra and XRD pattern before and after MF-SBC adsorption
图 12 MF-SBC吸附前后的FT-IR和吸附后的XRD图谱Figure 12. FT-IR spectra and XRD pattern before and after MF-SBC adsorption由图13(a)可以看出,吸附前后MF-SBC中Mg元素的特征峰分别在1 303.26 eV和1 304.42 eV处,归属于不同的Mg形态。吸附前位于1 303.26 eV处的特征峰表明Mg以金属氧键的方式,即氧化镁的形式存在;而反应之后在1 304.42 eV处的特征峰则归属于Mg的非金属形态,表明镁离子参与反应生成了鸟粪石沉淀。由图13(b)可见,不同形态的磷所处的结合能不同,吸附后MF-SBC的P2p峰可分为133.48 eV和134.28 eV 2个单峰,分别是鸟粪石沉淀表面的P=O和P—OH官能团。
3. 结论
1)通过煅烧法,成功合成了磁性污泥基生物炭MF-SBC,其表面含有大量的氧化镁颗粒,比表面积为58.99 m2·g−1,饱和磁化强度为52.48 emu·g−1。
2)在MF-SBC投加量为0.3 g·L−1,溶液初始pH为9时,MF-SBC同步吸附氨氮和磷酸盐的吸附量最大,分别为103.12 mg·g−1和205.07 mg·g−1,且吸附过程符合准二级动力学方程。Ca2+、Na+、SO42− 3种离子对磷酸盐的吸附无影响,而Ca2+和SO42−对氨氮的吸附有抑制作用。
3)在初始溶液pH=9的条件下,MF-SBC对水溶液中氨氮和磷酸盐同步吸附的作用机制包括表面吸附、离子交换和鸟粪石沉淀,其中鸟粪石沉淀为主要作用。
-
图 4 吸附前后MF-SBC脱吸附曲线和孔径变化
Figure 4. N2 adsorption and desorption curves and change in pore diameter of MF-SBC before and after adsorption
图 5 溶液初始pH对MF-SBC同步吸附氮磷的影响和MF-SBC的Zeta电位
Figure 5. Effect of initial pH on the adsorption capacity and zeta potential of MF-SBC
图 6 投加量对MF-SBC同步吸附氮磷的影响
Figure 6. Effect of dosage on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 7 接触时间对MF-SBC同步吸附氮磷的影响
Figure 7. Effect of the contact time on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 8 MF-SBC同步吸附氮磷的动力学拟合
Figure 8. Kinetic fitting of the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 9 共存离子对MF-SBC同步吸附氮磷的影响
Figure 9. Effects of coexisting ions on the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
图 10 MF-SBC在循环实验中的回收率以及对氨氮和磷酸盐的吸附容量
Figure 10. Recovery rate and adsorption capacity to ammonia nitrogen and phosphate by MF-SBC in cycle experiment
图 12 MF-SBC吸附前后的FT-IR和吸附后的XRD图谱
Figure 12. FT-IR spectra and XRD pattern before and after MF-SBC adsorption
表 1 MF-SBC同步吸附氨氮和磷酸盐的详细实验条件
Table 1. Experimental conditions for the simultaneous adsorption of nitrogen and phosphate by MF-SBC
影响因素 时间/min 氨氮质量浓度/(mg·L−1) P质量浓度/(mg·L−1) pH 共存离子 MF-SBC质量浓度/(g·L−1) pH 720 160 80 3~11 — 0.3 MF-SBC投加量 720 160 80 9 — 0.1~0.5 共存离子 720 160 80 9 Ca2+、Na+、SO42− 0.3 反应时间 1~720 160 80 9 — 0.3
下载: 导出CSV
表 2 MF-SBC同步吸附氮磷的动力学拟合参数
Table 2. Kinetic parameters for the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
污染物 准一级动力学 准二级动力学 Qe /(mg·g−1) k1/ min−1 R2 Qe/(mg·g−1) k1/(g·(mg·min)−1) R2 磷 168.44 0.0127 0.79 203.65 0.0012 0.99 氨氮 83.86 0.013 0.77 101.34 0.0017 0.99
下载: 导出CSV
表 3 MF-SBC同步吸附氮磷的颗粒内扩散模型参数
Table 3. Intra particle diffusion model parameters for the simultaneous adsorption of nitrogen and phosphorus by MF-SBC
污染物 S1 S2 S3 K R2 K R2 K R2 磷 11.57 0.95 25.07 - 0.83 0.92 氨氮 7.56 0.98 2.43 - 0.28 0.92
下载: 导出CSV
-
[1] 赵玉芬. 磷化学与化工进展[J]. 中国科学:化学, 2010, 40(7): 801. [2] 孙理密, 翟纪学, 张德清, 等. 高氮磷有机食品废水处理工程实例分析[J]. 工业水处理, 2022, 42(1): 171-174. [3] 聂坤, 杨成建, 李志华, 等. 鸟粪石结晶-絮凝沉淀同步法回收养殖场废水的氮磷[J]. 水处理技术, 2022, 48(7): 38-42. [4] XIE F, WU F, LIU G, et al. Removal of phosphate from eutrophic lakes through adsorption by in situ formation of magnesium hydroxide from diatomite[J]. Environmental Science & Technology, 2014, 48(1): 582-590. [5] ZENG, F Z, ZHAO Q L, JIN W B, et al. Struvite precipitation from anaerobic sludge supernatant and mixed fresh/stale human urine[J]. Chemical Engineering Journal, 2018, 344: 254-261. doi: 10.1016/j.cej.2018.03.088 [6] 李亮, 王德汉, 邹璇. 曝气在沉淀法回收沼气发酵液氮磷中的作用[J]. 农业工程学报, 2010, 26(1): 313-318. [7] 张琪, 赵首萍, 叶雪珠, 等. 鸟粪石结晶法回收氮磷的影响因素研究[J]. 科技通报, 2015, 31(7): 237-244. [8] 霍守亮, 席北斗, 刘鸿亮. 磷酸铵镁沉淀法去除与回收废水中氮磷的应用研究进展[J]. 化工进展, 2017, 26(3): 371-376. [9] 杨明珍, 包震宇, 师晓春, 等. 鸟粪石沉淀法处理沼液实验研究[J]. 工业安全与环保, 2016, 37(3): 31-32. [10] BARBOSA S G, PEIXOTO L, MEULMAN B, et al. A design of experiments to assess phosphorous removal and crystal properties in struvite precipitation of source separated urine using different Mg sources[J]. Chemical Engineering Journal, 2016, 298: 146-153. doi: 10.1016/j.cej.2016.03.148 [11] 吴彦霖, 周荣敏. MAP法与沸石吸附组合工艺的脱氮除磷实验研究[J]. 中国环保产业, 2015(2): 45-48. [12] HUANG H, XIAO D, ZHANG Q, et al. Removal of ammonia from landfill leachate by struvite precipitation with the use of low-cost phosphate and magnesium sources[J]. Journal of Environmental Management, 145: 191-198. [13] 张记市, 王玉松. 鸟粪石结晶法回收垃圾渗滤液氨氮研究[J]. 环境工程学报, 2009, 3(11): 2017-2020. [14] ZHANG J S, WANG Q Q. Sustainable mechanisms of biochar derived from brewers' spent grain and sewage sludge for ammonia-nitrogen capture[J]. Journal of cleaner production, 2016, 112: 3927-3934. doi: 10.1016/j.jclepro.2015.07.096 [15] HALL K E, CALDERON M J, SPOKAS K A, et al. Phenolic acid sorption to biochars from mixtures of feedstock materials[J]. Water Air & Soil Pollution, 2014, 225(7): 2031. [16] FENG Y, XU Y, YU Y, et al. Mechanisms of biochar decreasing methane emission from Chinese paddy soils[J]. Soil Biology & Biochemistry, 2012, 46(1): 80-88. [17] 李时琛, 方海旭, 李俊青. 菜叶生物质炭的磷酸活化制备及其吸附性能研究[J]. 化工管理, 2018(1): 80-81. [18] BOESCH D F, BURRESON E, DENNISON W, et al. Factors in the decline of coastal ecosystems[J]. Science, 2001, 293(5535): 1589-1691. [19] ENDERS A, HANLEY K, WHITMAN T, et al. Characterization of biochars to evaluate recalcitrance and agronomic performance[J]. Bioresource Technology, 114: 644-653. [20] OZCAN A, OZCAN A S, TUNALI S, et al. Determination of the equilibrium, kinetic and thermodynamic parameters of adsorption of copper (II) ions onto seeds of Capsicum annuum[J]. 2005, 124: 200-208. [21] 杨奇亮, 吴平霄. 改性多孔生物炭的制备及其对水中四环素的吸附性能研究[J]. 环境科学学报, 2019, 39(12): 3973-3984. [22] LIU J W, JIANG J G, AIHEMAITI A, et al. Removal of phosphate from aqueous solution using MgO-modified magnetic biochar derived from anaerobic digestion residue[J]. Journal of Environmental Management, 2019, 250: 109438. doi: 10.1016/j.jenvman.2019.109438 [23] CUI X, HAO H, ZHANG C, et al. Capacity and mechanisms of ammonium and cadmium sorption on different wetland-plant derived biochars[J]. Science of the Total Environment, 2016, 539: 566-575. doi: 10.1016/j.scitotenv.2015.09.022 [24] LYU H H, GAO B, HE F, et al. Effects of ball milling on the physicochemical and sorptive properties of biochar: Experimental observations and governing mechanisms[J]. Environmental Pollution, 2018, 233: 54-63. doi: 10.1016/j.envpol.2017.10.037 [25] YAN H, SHIH K. Effects of calcium and ferric ions on struvite precipitation: A new assessment based on quantitative X-ray diffraction analysis[J]. Water Research, 2016, 95: 310-318. doi: 10.1016/j.watres.2016.03.032 [26] CHENG S, ZHAO S, GUO H, et al. High-efficiency removal of lead/cadmium from wastewater by MgO modified biochar derived from crofton weed[J]. Bioresource Technology, 2022, 343: 126081. doi: 10.1016/j.biortech.2021.126081 [27] LI A, XIE H, QIU Y, et al. Resource utilization of rice husk biomass: Preparation of MgO flake-modified biochar for simultaneous removal of heavy metals from aqueous solution and polluted soil[J]. Environmental Pollution, 2022, 310: 119869. doi: 10.1016/j.envpol.2022.119869 [28] WU J W, WANG T, WANG J, et al. A novel modified method for the efficient removal of Pb and Cd from wastewater by biochar: Enhanced the ion exchange and precipitation capacity[J]. Science of the Total Environment, 2021, 754: 142150. doi: 10.1016/j.scitotenv.2020.142150 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4619
- HTML全文浏览数: 4619
- PDF下载数: 70
- 施引文献: 0
