

-
近年来,抗生素等药品和个人护理品(pharmaceutical and personal care products, PPCPs)在地表水和地下水环境样品中不断被检出,这说明其在水生生态系统中存在越来越广泛的分布[1-4],对水生生物和人类健康构成潜在威胁[5-6]。作为PPCP的典型代表之一,磺胺甲恶唑(sulfamethoxazole, SMX)是一种广泛应用的人、兽两用的磺胺类抗生素,由于其消费量巨大且不易降解,导致其在环境水体中广泛赋存[7],从而存在诱发细菌产生抗生素耐药基因的风险[8-9]。因此,开发SMX的有效降解方法很有必要。
高级氧化工艺(advanced oxidation processes, AOPs)是一种通过催化产生自由基等强活性氧物质(reactive oxygen species,ROS)来处理有机污染物的有效方法[10-13]。硫酸根自由基(SO4·−)由于具有较高的氧化还原电位(2.5~3.1 eV)、较长的半衰期(30~40 μs)和对芳烃环污染物具有较强的选择性氧化能力,近年来基于硫酸根自由基的高级氧化(SO4·−-based AOPs, SR-AOPs)技术在降解毒性有机污染物的研究中备受关注[14-15]。过一硫酸盐(peroxymonosulfate, PMS),可通过光化学、声化学和热活化、过渡金属(如钴、铁、银和铜)、碳质材料、电化学活化、碱性条件和氧化剂(如臭氧、过氧化氢、和过氧化钙)等多种途径破坏(氢化)过氧化键(O-O)活化生成SO4•−[15-16]。其中过渡金属钴(Co)活化PMS是目前产生SO4·−的最为高效的一种方法。有研究表明,Co(Ⅱ)/PMS体系在中性pH条件下的催化性能优于传统的芬顿(Fenton)反应,且试剂用量更少[17]。然而,直接使用Co(Ⅱ)作为均相催化剂通常存在可循环性低、Co(Ⅱ)污泥排放存在潜在风险等缺点[11]。为提高催化活性、稳定性和循环使用性能,通常采用其他载体负载Co来制备多相催化剂。常见的载体有碳材料、粘土、沸石和其他金属氧化物等。
近年来,基于静电纺丝工艺制备的碳纤维(carbon nanofibers, CNFs)材料因其制备方法简单、材料比表面积大、电子传输性能好引起了众多研究者的关注[18-19]。CNFs具有较高的长径比,有利于电子传输和反应物扩散,以此为载体可表现出优异的催化活性。目前,钴掺杂碳纳米纤维作为SR-AOPs的高效多相催化剂已有报道[18, 20]。LIN等[21]和ZHANG等[22]发现,负载Co与其他金属元素(Fe和Ag)的双金属纳米颗粒碳纳米纤维,在低剂量下表现出比单金属催化剂更好的性能,且能减少金属离子的浸出。然而,Co/Fe体系通常表现出高磁性和团聚,因此,不可避免地降低了催化活性[23-24];Co/Ag体系应用成本较高,不利于催化剂的广泛使用。Ti也是一种过渡金属,其氧化物具有良好的催化性,在水溶液中可产生氧自由基和羟基自由基等活性基团,当与Co复合时,有望进一步改善Co基催化剂的相关性能。
因此,本研究采用静电纺丝技术,以聚丙烯腈(PAN)为前驱体,适用一步法制备了Co/TiO2@CNFs复合纳米纤维薄膜,通过调整Co和TiO2的复合比例,优化了复合纤维膜的最佳合成工艺;以磺胺甲恶唑(SMX)作为目标污染物,考察了Co/TiO2@CNFs活化PMS降解SMX的效能,且探讨了可能的降解途径及机理。
-
磺胺甲恶唑(SMX)、聚丙烯腈(PAN, FW为150 000 Da)、N, N-二甲基甲酰胺(DMF)和过硫酸氢钾复合盐(Oxone, KHSO5·0.5KHSO4·0.5K2SO4)、对苯醌(p-BQ)、糠醇(FFA)、L-组氨酸(L-histidine)均购自Sigma-Aldrich公司(St. Louis, MO, USA);六水硝酸钴(Co(NO3)2·6H2O)、二氧化钛(TiO2)、氢氧化钠(NaOH)、硫酸(H2SO4)、叔丁醇(TBA)和甲醇(methanol)均购自中国医药化学试剂有限公司(上海);所用材料均为分析纯。高效液相色谱(HPLC)级乙腈(acetonitrile)、甲醇(methanol)和甲酸(methanoic acid)由J.T. Baker (Phillipssburg, NJ, USA)公司购得。使用Millipore Milli-Q Direct 8/16净水系统(中国长沙)制备去离子水(<18.3 MΩ·cm−1)。
-
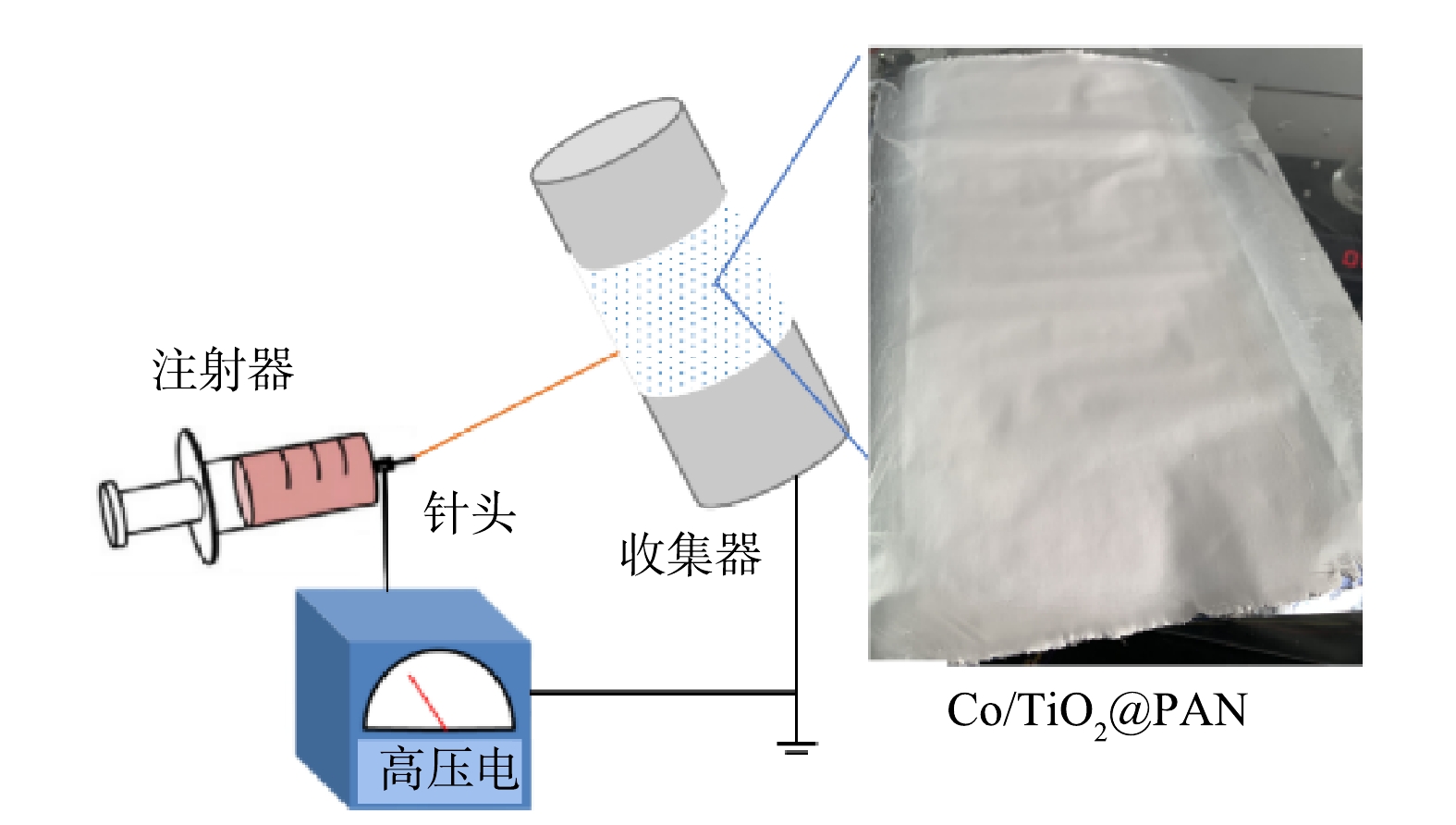
将2.60 g PAN聚合物粉末溶解在25 g DMF溶液中,室温下磁力搅拌至溶液清澈透明。加入0.032 g TiO2,超声分散4 h后添加0.55 g Co(NO3)2·6H2O,充分混合后配得纺丝前驱液。如图1所示,将前驱液转移至注射器,配置1.2 mm×38 mm规格不锈钢针头,在0.4 mL·h−1推进速率、16 KV高压电、6 cm间距条件下进行静电纺丝,同步制备Co/TiO2@PAN纳米纤维膜。对上述所制备的膜进行热处理:在240 ℃空气中煅烧1 h后再在N2保护下600 ℃煅烧3 h。纳米纤维被标记为X Co/TiO2@CNFs,其中X代表钛的质量千分比,本研究采用的催化剂1 Co/TiO2@CNFs简称为Co/TiO2@CNFs。
-
催化剂活化PMS降解SMX实验在100 mL玻璃烧杯中以批实验进行。各因素实验条件为:催化剂质量浓度(0、0.05、0.10和0.20 g·L−1),PMS浓度(0、0.25、0.5、1.0和2.0 mmol·L−1),水温(15、20、25、30和40 ℃),pH(3、5、7、9、11和未调节值5.8)进行性能测试。采用水浴进行温度控制,用0.1 mol·L−1 H2SO4和NaOH溶液调节溶液pH。催化降解实验:将5 mg催化剂投加到50 mL SMX溶液(40 μmol·L−1)中,搅拌3 min至均匀分散,后投加适量的PMS至所需浓度。水样定时采样,经0.22 μm滤膜过滤后再进行甲醇淬灭反应,最终鉴定SMX及降解产物。淬灭实验:先在SMX溶液中分别加入甲醇(MeOH)、叔丁醇(TBA)、对苯醌(p-BQ)、糠醇(FFA)或L-组氨酸(L-histidine)等淬灭剂后采用相同的催化降解步骤。循环测试实验:将使用过的催化剂,经膜过滤后用去离子水和甲醇各洗涤3次,60 ℃烘干待用。
-
采用扫描电子显微镜(SEM, Hitachi s-4800, Japan)和透射电子显微镜(TEM, Feitecnai G2 F30, USA)表征材料的形貌和微观结构;采用能量色散X射线光谱仪(NS7, Thermo Fisher Scientific, USA)与TEM联用表征材料的元素组成;采用X射线衍射仪(D8 Advance, Germany)进行材料的晶格分析;采用傅里叶红外光谱仪(Nexus-470, Thermo Nicolet, USA)表征材料表面官能团化学键结构;采用规格/型号为*inVia Qontor/ inVia Qontor仪器进行拉曼分析;采用热重分析仪(TA Instruments, USA)进行材料热重分析;采用Autosorb-iQ自动气体吸附分析仪(Quanta Chrome Instruments, USA)测量材料的比表面积和孔径分布;采用纳米粒度电位仪(Zetasizer Nano ZS, Malven Instruments, UK)进行Zeta电位分析;采用X射线光电子能谱仪(ESCALAB 250Xi, Thermo Fisher Scientific)表征材料的化学组成和化学状态。
采用高效液相色谱仪(HPLC, Agilent 1260, USA),配置XDB-C18柱(150 mm × 4.6 mm, i.d., 5 μm particle,Waters, USA)和VWD检测器分析SMX浓度。检测条件设置为:流动相采用甲醇和水体积比为55∶45;流速采用0.8 mL·min−1;柱温控制为35 ℃;波长为275 nm。使用电感耦合等离子体质谱仪(ICP-MS, Agilent 7700, USA)测定溶液中金属离子浓度。采用高效液相色谱-电喷雾电离串联质谱(HPLC-ESI-MS/MS)结合TSQ Quantum Access MAX设备(Thermo, USA),配备反相C18分析柱(100 mm × 2.1 mm, 5 μm, Thermo, USA) 对SMX降解中间产物进行鉴定,样品在分析前用MCX (Anpel, China)固相萃取柱提取。采用从50到400 m·z−1全扫描质量方法下的正模式电喷雾电离(ESI +)分析产物中间体,柱温度30 ℃,流量0.15 mL·min−1,流动相是A(乙腈)和B(0.1%甲酸水)。用电子顺磁共振波谱仪(Bruker ESP300E, Germany)进行活性物种鉴别。
活化PMS降解SMX参数采用假一级动力学方程(式(1))进行分析。采用阿伦尼乌斯(Arrhenius)方程(式(2))研究kapp与不同温度之间的关系。
式中:Ct为t时刻污染物浓度,μmol·L−1;C0为污染物初始浓度,μmol·L−1;kapp为表观动力学常数,min−1;t为时间,min。
式中:A为指前因子,g·(mg·min)−1;Ea为SMX降解的活化能,kJ·mol−1;R为通用气体常数,8.314 J·(mol·K)−1;T为溶液的温度,K。
-
通过SEM、HRTEM、TEM、HAADF-STEM和元素映射对Co/TiO2@CNFs的形貌进行了表征(图2)。由图2(a)可以发现,所制备的材料是平均直径为200~300 nm、相互交织形成开放网状结构的多孔纤维。图2(b)显示,纤维表面负载了颗粒状固体。由图2(c)可见,在材料表面存在晶格条纹间距为0.240~253 nm的钴氧化物(Co3O4),但分布不均匀。在图2(d)中可以看到纤维表面有很多亮点分布。进一步采用高角环形暗场扫描透射原子成像(HAADF-STEM)可发现:这些亮点以亮斑呈现(图2(e))。元素映射分析结果表明(图2(f)~(k)),这些亮斑与Co的分布完美对应, 而O的分布在对应Co的位置也明显明亮。综上所述,由于Ti、N及C在纤维表面分布均匀,因此,不难推测,除了一部分与Ti结合的O(TiO2)及与N结合的O(Co(NO3)2)在表面均匀分布外,还有一部分很可能在局部与Co形成了氧化物。图3(a)是不同成分含量的Co/TiO2@CNFs的XRD测试结果,随着TiO2含量的增加,在(101)、(103)、(004)、(112)、(200)、(105)、(211)、(204)、(116)、(220)、(215)(JCPDS#99-0008)处的TiO2特征衍射峰逐渐增加,这进一步证实了TiO2成功复合在CNFs上。XRD未检测到显著的Co特征峰,可能是材料中Co的结晶度较低,或其特征峰较弱而被TiO2特征峰掩盖。Co@CNFs和X Co/TiO2@CNFs的拉曼光谱如图3(b)所示,在674 cm−1附近的拉曼波段代表晶体Co3O4的A1g模态[25],证实该晶体成功复合到CNFs上。
-
根据拉曼光谱(图3(b)),5种碳材料在1 350 cm−1和1 575 cm−1附近都有显著的特征峰,即D峰和G峰。并且,Co@CNFs、0.5Co/TiO2@CNFs、1Co/TiO2@CNFs、5Co/TiO2@CNFs和10Co/TiO2@CNFs的ID/IG值分别为2.85、3.00、3.33、2.23和2.45,以1Co/TiO2@CNFs的ID/IG值最高,5Co/TiO2@CNFs最低。ID/IG越大,表明晶体缺陷越多,越有利于催化氧化。
相关材料的热重分析(TGA)结果如图3(c)所示。与CNFs相比,Co/TiO2@CNFs和Co/CNFs在100~300 ℃内表现出持续失重。当温度为300~400 ℃时,催化剂的失重更加明显,这可能是由该温度范围内挥发性成分的脱除、脱氢和环化所致[18]。由于Co和TiO2的添加会引起CNFs的缺陷增加,从而导致该温度范围内的脱氢、环化等化学反应增加,因此,表现出比CNFs更明显的失重。在450~800 ℃内,Co/TiO2@CNFs热稳性比Co@CNFs更好,说明掺杂TiO2可提高Co@CNFs的稳定性。
对比Co@CNFs和Co/TiO2@CNFs的FTIR表征结果(图3(d))可发现新增O—H在3 425 cm−1处及C—H在2 919 cm−1处的伸缩振动特征吸收峰;而苯环C=C在1 588 cm−1处以及C—C在1 266 cm−1处的伸缩振动特征吸收峰得到强化。说明添加TiO2后,其碳骨架形成了石墨平面网状层稳定结构,表面含有O—H官能团。图3(e)为Co/TiO2@CNFs 在不同pH下的Zeta电位变化。可见Co/TiO2@CNFs的pHPZC为6.7。因此,当pH低于6.7时,催化剂的表面电荷为正电荷,反之为负电荷。
Co@CNFs和XCo/TiO2@CNFs的N2吸附/脱附等温线分析结果如表1所示。其中1Co/TiO2@CNFs的比表面积为97.15 m2·g−1,总孔容为0.092 cm3·g−1,t-图法微孔为0.018 cm3·g−1,介孔体积是0.074 cm3·g−1,BJH孔径确定为3.93 nm。上述结果表明,1Co/TiO2@CNFs在5种催化剂中具有最大的比表面积和总孔容。
通过以上表征分析可知,1Co/TiO2@CNFs相比其他催化剂缺陷度更多、比表面积更大、总孔容更大,理论上应具有更好的催化性能。
-
图3(f)为XCo/TiO2@CNFs对SMX的吸附和催化降解效果。前20 min未添加PMS,SMX未出现明显下降,说明该类催化剂对水中的SMX吸附作用几乎可以忽略。20 min后向溶液中加入PMS,SMX浓度迅速下降,60 min时Co@CNFs、0.5Co/TiO2@CNFs、1Co/TiO2@CNFs、2Co/TiO2@CNFs、5Co/TiO2@CNFs、10Co/TiO2@CNFs对SMX的降解率分别为82.9%、92.6%、93.3%、91.2%、86.3%、73.6%。上述结果表明,当催化剂中TiO2添加量从0.5增至5时,催化剂对SMX的降解率分别提升了11.7%、12.5%、10.0%、4.1%。当TiO2添加量为1时,即1Co/TiO2@CNFs活化PMS降解SMX的效果最好。根据之前的表征结果推测,可能原因如下:1) 1Co/TiO2@CNFs具有最大的比表面积,而催化剂的比表面积越大,活性位点越多;2) 1 Co/TiO2@CNFs中碳晶体缺陷度最大,有利于电子转移可促进PMS活化;3) FTIR光谱表明Co/TiO2@CNFs的表面有大量OH基团,可促进催化剂表面Co-OH配合物的形成,从而促进了PMS的多相活化[26-27]。从XPS O1s精细谱对比分析(图4(a))中可以看出,Co/TiO2@CNFs的C-O/Co-O多于Co@CNFs。结合图4(c)~(d),反应后C-O/Co-O有所降低,这可以看出表面Co-OH配合物在降解反应中发挥了重要作用。当TiO2添加量为10时,其催化性能反而略低于Co@CNFs,表明过多的TiO2会导致复合催化剂性能降低。这可能是由于添加过多TiO2后,存在团聚或包覆等问题导致Co3O4作用降低。因此,后续实验催化剂均选用1Co/TiO2@CNFs。
-
催化剂投加量对SMX降解的影响如图5(a)所示。在PMS浓度为1 mmol·L−1, 反应温度为 25 ℃条件下,当仅添加PMS时,SMX在50 min内几乎无法降解,说明PMS难以自分解去除SMX。当Co/TiO2@CNFs投加量从0.05 g·L−1增至0.2 g·L−1,对应的由式(1)求得的降解动力学常数kapp从0.065 8 min−1提升至0.206 4 min−1。这是因为催化剂越多,可提供的活化PMS位点越多,从而可提升SMX的降解率。
图5(b)反映了PMS剂量对SMX降解的影响。可见,反应动力学和SMX的降解程度受到PMS浓度变化的影响。未添加PMS时,SMX的浓度无明显下降,表明催化剂无吸附效果,这与上述结论一致。当PMS剂量由0.25 mmol·L−1增加到2 mmol·L−1,反应动力学常数 kapp由0.061 8 min−1增长到0.157 6 min−1。这可能是由于PMS剂量越多,PMS与催化剂表面的碰撞概率增大并产生更多活性物种,从而提升了降解效率。
图5(c)反映了温度对SMX降解效果的影响。当温度从15 ℃增至40 ℃时,对应kapp从0.045 4 min−1升至0.349 1 min−1。此结果表明,温度升高可加速SMX的催化降解速率。这是因为温度升高增加了溶液中分子的碰撞概率,促进了PMS的活化,增加活性物质的产生。采用阿伦尼乌斯(Arrhenius)方程(式(2))研究了kapp值与温度之间的关系。由图5(c)中的插图可见,数据点的线性回归拟合较好,计算可得Ea为63.23 kJ·mol−1。
图5(d)反映了pH对SMX降解的影响。SMX溶液的自然pH为5.8,当pH降至3和升高至11时,SMX的降解kapp分别从0.139 6 min−1降为0.071 8 min−1和0.011 0 min−1。可见,SMX降解的最佳pH为5.8~9,而酸性和碱性过强均不利于SMX的降解。其原因归为以下4点。1)在强酸条件下,·OH和SO4·−可被H+灭活(式(3)~式(4))[28-29],从而抑制了PMS的活化和SMX的降解。酸性条件也可抑制CoOH+络合物的形成,从而降低了SO4·−的生成[8]。2)PMS在酸性或中性条件下主要存在形式是HSO5−,活化生成的主要物质是SO4·−。而强碱性条件下主要存在形式是SO52−,其催化活性较低[11]。3) SMX在不同的pH下分别呈现质子化、非质子化和去质子化形式(阳离子、中性和阴离子形式),pKa1为1.6,pKa2为5.7[1, 30]。当pH为3.0时,SMX以非质子化SMX为主;当pH增加到7.0时,SMX主要存在形式为去质子化SMX。Zeta电位结果表明,Co/TiO2@CNFs的pHpzc为6.7(图3(e))。因此,在弱酸性pH下,带负电的去质子化SMX与带正电的催化剂表面的亲和力更强,催化反应效率能得到提升[18];而当碱性pH时,催化剂表面带负电,不利于PMS活化反应的发生。4)过于碱性环境会导致催化剂表面形成覆盖表面活性位点的CoOH+络合物,从而增加SMX与催化剂之间的静电斥力。
图6反映了共存的无机离子Cl−、F−、HCO3−和H2PO4−对SMX降解的影响。如图6(a)所示,当Cl−为0~30 mmol·L−1时,SMX在3 min内即可完全降解。这可能是Cl−导致SO4·−和·OH产生2.4 V高氧化还原电势的氯自由基Cl•[31-32],生成的Cl·还可与Cl−反应生成Cl2·−(式(5)~式(8))[32-33]。此外,PMS与Cl−反应可生成氧化性氯离子,如HOCl和Cl2 (式(9)~式(10)),Cl·与水反应可生成·OH降解SMX(式(11))[28]。图6(b)表明,F−的添加与Cl−对SMX降解具有相似的促进影响,这可能是因为两者具有相似的卤素元素化学特征。
由图6(c)可知,当HCO3−浓度增加到30 mmol·L−1时,SMX降解反应速率明显加快。这可能是HCO3−的缓冲能力抑制了H+对SO4·−和·OH的捕获[11]。由图6(d)可见,当NaH2PO4为0~30 mmol·L−1时,SMX的降解率有所下降。这可能是H2PO4−和SO4·−或·OH反应形成H2PO4·(式(12)~式(13)),但在50 min时的降解率仍然超过90%。此外,H2PO4−由于与固体表面的强亲和力,易在催化剂表面形成络合物[8,34],这可能导致Co/TiO2@CNFs上的活性位点降低。
-
通过循环实验研究了Co/TiO2@CNFs的可重复利用性。如图7所示,在5次循环降解实验中,Co/TiO2@CNFs对SMX的降解率分别为≥99%、99%、93%、91%、89%,对应降解动力学常数kapp分别为0.139 6、0.108 2、0.049 0、0.042 2、0.039 4 min−1。虽然SMX的去除率随着Co/TiO2@CNFs使用频率的增加而有所下降,但经5次循环后的SMX去除率仍保持在较高水平。这可能归因于以下2点。1) 材料表面部分活性位点被降解中间体屏蔽。如图4(e)所示,催化剂表面吡咯型N及C-N-H含量在反应后有所上升,表明催化剂表面活性位点可能存在降解中间体屏蔽。2)材料表面的部分Co金属泄漏。经ICP-MS测试可知,Co@CNFs和Co/TiO2@CNFs在催化反应后溶液中Co泄漏量分别为0.40 mg·L−1和0.36 mg·L−1,均低于1.0 mg·L−1,满足《城镇污水处理厂污染物排放标准》(GB 18918-2002)及《地表水环境质量标准》(GB 3838-2002)的规定要求。该泄漏量也与BAO等[18]制备的5Co/CNF和8Co/CNF在反应120 min后Co泄漏量(0.31 mg·L−1和0.52 mg·L−1)和WANG等[35]制备的Co/Co9S8@N-S-OC在反应250 min后Co泄漏量(0.52 mg·L−1)相当,其泄漏量相对不高,说明稳定性较好,且Co/TiO2@CNFs相比Co@CNFs性能更优。
-
通过液相和质谱联用鉴定出可能的中间产物共10种。根据质荷比(m/z)推测其化学分子式和结构式,进一步分析SMX在Co/TiO2@CNFs活化PMS体系中降解的可能转化途径。中间产物用P和质荷比组合的方式来表述。如图8所示,在路径A中,PMS中的Na+可以与SMX结合形成P276;在路径B中,通过ROS使S—N键断裂而产生P173和P99,即4-氨基-苯磺酸盐和3-氨基-5-甲基-异恶唑[11,36]。此外,自由基可将S—N断裂也可能产生P156和P99[8]。P173通过羟基化作用和氨基的丢失可被进一步氧化生成P143[36]。在路径C中,SMX中异恶唑环氧化开环后形成P217,S—N键进一步发生断裂,产生P156或P173,P156可进一步氧化生成P125。路径D是SMX苯环上的氨基被自由基氧化生成P267和亚硝基衍生物[37],P267可以进一步氧化得到其他小分子产物。
-
通过ESR分析和淬灭实验对Co/TiO2@CNFs活化PMS降解SMX反应中的主要活性物种进行了鉴定。利用5,5 -二甲基-1-吡咯烷N-氧化物(DMPO)和2,2,6,6-四甲基哌啶(TEMP)作为自旋捕获剂,分别得到了SO4·−、·OH、O2·−和1O2的ESR分析特征峰,如图9(a)~(c)所示。为进一步确定哪种活性氧物种是SMX降解的主要活性物种,采用TBA、甲醇、p-BQ、L-组氨酸和FFA分别作为·OH、SO4·−、O2·−和1O2的淬灭剂,结果显示,60 min时SMX降解率分别为89.5%、60.5%、49.0%、12.9%、4.3%,而未添加淬灭剂的对照组降解率为99.2%。上述结果说明,1 mol·L−1 TBA的添加对活化PMS降解SMX效率的影响较小,而1 mol·L−1甲醇的添加进一步抑制SMX的降解效率,表明实验过程中产生了少量·OH和SO4·−。5 mmol·L−1 p-BQ的添加进一步影响了PMS活化降解SMX,表明溶液中存在一定的O2·−。100 mmol·L−1 L-组氨酸和100 mmol·L−1FFA的添加几乎完全抑制了SMX降解,表明实验过程中产生了大量1O2。综上所述,4种活性物种均有产生,其中1O2在Co/TiO2@CNFs活化PMS过程起到了主导作用。此外,溶液中可能存在的机理反应如式(14)~式(25)[34, 38-41]所示。
为了更深入地解释反应机理,采用XPS分析了Co/TiO2@CNFs在反应前后的元素组成和化学价态。如图4(f)所示,在796.5 eV和780.8 eV的特征峰分别对应于Co2p1/2和Co2p3/2。其中,2个振荡峰分别位于802.7 eV和786.4 eV,Co2p3/2 (780.7 eV)和Co2p1/2 (796.4 eV)的峰属于Co(Ⅲ),Co2p3/2 (782.5 eV)和Co2p1/2 (797.8 eV)的峰属于Co(Ⅱ)。催化反应后Co(Ⅲ)的相对比例略有增加,而Co(Ⅱ)的相对比例下降,说明催化剂在Co/TiO2@CNFs表面上形成了Co(Ⅲ)[17]。这一结果表明在降解过程中发生了氧化还原反应。如式(26)~式(28)所示[22],由于Co(Ⅱ)直接导致SO4·−自由基的生成,因此,催化剂表面的Co(Ⅱ)可以显著促进界面催化反应。有研究表明,催化剂表面生成Co-OH络合物是活化PMS最有效的物种,是反应限速步骤[17]。根据前面的FTIR分析结果,添加TiO2后复合催化剂表面有大量OH基团,可以促进催化剂表面Co-OH络合物的形成,并可强化SMX的降解效率。这说明TiO2强化的Co(Ⅱ)-Co(Ⅲ)-Co(Ⅱ)氧化还原循环参与了催化降解反应。
综上所述,提出了SMX在Co/TiO2@CNFs活化PMS体系中降解的反应机理(图10)。静电纺丝纤维材料表面的Co(Ⅱ)-Co(Ⅲ)-Co(Ⅱ)氧化还原循环活化PMS产生SO4·−和·OH物质,并进一步转化成O2·−和1O2,4种活性物种共同促进SMX的降解。而在Co(Ⅱ)-Co(Ⅲ)-Co(Ⅱ)氧化还原循环当中,TiO2促进了催化剂表面Co-OH的形成,从而提升了催化反应效率。
-
1)通过静电纺丝工艺和热处理技术直接制备得到Co/TiO2@CNFs复合碳纳米纤维催化剂,该材料具有多孔网状结构、较好的稳定性和重复利用性,并能与PMS耦合实现水中SMX的高效降解。在材料投加量为0.1 g·L−1,PMS投加量为1 mmol·L−1条件下,50 min后SMX降解率达99.9%,复合催化剂的最佳pH为5.8~9。
2) TiO2的添加相比于仅仅负载Co的纳米纤维,其催化能力和材料稳定性能均有提高,其中添加比例为0.1%时,Co/TiO2@CNFs具有最大的比表面积、缺陷度,且TiO2添加后可促进催化剂表面Co-OH的形成,因此其催化降解SMX的效率最高。
3)该复合催化剂对SMX进行了有效降解,并根据10种检测到的中间产物推断出其降解过程的4条反应路径。Co/TiO2@CNFs对SMX的高效降解主要通过高效生成1O2和TiO2促进催化剂表面Co-OH的形成来共同实现。
载钴钛碳纳米纤维活化过一硫酸盐降解磺胺甲恶唑的效能及机理分析
Degradation efficiency and mechanism of sulfamethoxazole by activation of peroxymonosulfate via cobalt titanium-loaded carbon nanofibers
-
摘要: 本研究采用静电纺丝和热处理技术制备了一种网状负载钴钛双金属纳米颗粒碳纳米纤维催化剂(Co/TiO2@CNFs),并通过SEM、TEM、HRTEM、HAADF-STEM、元素映射、XRD、氮气吸脱附、TGA、拉曼光谱、FTIR、XPS、Zeta电位等手段对催化剂进行了表征和分析,通过批式降解实验优化了二氧化钛(TiO2)最佳负载量,并研究了催化剂投加量、PMS投加量、温度、pH、共存离子(Cl−、F−、HCO3−和H2PO4−)等因素对磺胺甲恶唑(SMX)降解性能的影响。结果表明,TiO2负载可提高催化剂的催化性能,TiO2与所制备催化剂的最佳质量比为0.1%;在催化剂投加量为0.1 g·L−1,PMS投加量为1 mmol·L−1时,Co/TiO2@CNFs在50 min内降解SMX≥99%;在pH为5.8~9内反应最佳;根据阿伦尼乌斯方程计算出反应体系的活化能为63.23 kJ·mol−1;催化剂在4种共存离子存在下表现出较好的催化性能;经5次循环后,Co/TiO2@CNFs催化降解性能仍保持较好。ESR分析和淬灭实验结果表明,4种活性物种(·OH、SO4·−、O2·−和1O2)参与到SMX的降解过程中,其中,1O2在Co/TiO2@CNFs活化PMS过程中起主导作用。Abstract: The catalytic activation of peroxymonosulfate (PMS) to generate free radicals has received a lot of attention in the environmental catalytic treatment of refractory pollutants. Electrostatic spinning and heat treatment procedures were used to manufacture a porous cobalt-titanium bimetallic nanoparticles supported carbon nanofibers catalyst (Co/TiO2@CNFs) in this research. SEM, TEM, HRTEM, HAADF-STEM, elemental mapping, XRD, nitrogen adsorption/desorption isotherms, TGA, Raman spectroscopy, FTIR, XPS, Zeta potential, and other methods were used to characterize the physical and chemical properties of catalysts. The optimal titanium dioxide (TiO2) loading capacity was explored by batch degradation experiment. The effects of catalyst dosage, PMS dosage, temperature, pH and coexisting ions (Cl−、F−、HCO3− and H2PO4−) on the degradation efficiency of sulfamethoxazole (SMX) were investigated. The results show that TiO2 supports could improve the catalytic performance of the catalyst, and the optimal mass ratio of TiO2 to catalyst was 0.1 %. When catalyst dosage was 0.1 g·L−1 and PMS dosage was 1 mmol·L−1, Co/TiO2@CNFs could degrade SMX≥99% within 50 minutes. The optimum pH value was 5.8-9. According to Arrhenius equation, the activation energy of the reaction system was 63.23 kJ·mol −1. The catalyst had a good catalytic performance under the condition of four coexisting ions. After 5 cycles, Co/TiO2@CNFs still maintained a good catalytic degradation performance. Four active species (·OH、SO4·−、O2·− and 1O2) were involved in the degradation of SMX by ESR analysis and quenching experiments, of which 1O2 played a dominant role in the Co/TiO2@CNFs activated PMS system. Finally, the possible pathway and reaction mechanism of SMX degradation in bimetallic co-catalytic system are proposed. The findings of this research provide a novel perspective on the application of advanced oxidation processes (AOPs) in environmental remediation.
-
Key words:
- peroxymonosulfate /
- electrospinning /
- carbon nanofibers /
- sulfamethoxazole /
- reaction mechanism.
-
多溴联苯醚(poly brominated diphenyl ethers, PBDEs)是一种广泛应用的添加型溴系阻燃剂,可经产品的生产、使用和处置回收等途径释放到环境中[1],世界范围的水体、土壤、沉积物、大气、动植物甚至人体中均可检出[2 − 4]. PBDEs脂溶性较强,容易在脂肪组织蓄积,并通过食物链放大,具有神经毒性[5]、肝毒性[6]、免疫毒性[7]、遗传毒性和内分泌干扰效应等[8],也是潜在的致癌物[9]. 2009年,斯德哥尔摩公约将含有4—7个溴原子的4种PBDEs列入持久性有机污染物名单. BDE-47是商用PBDEs的主要成分,海洋环境中,BDE-47是生物利用度最高的低溴代PBDEs之一,约占PBDEs总量的70%,在水体和沉积物中浓度高(ND—pg·L−1/ND-ng·g−1),生物毒性比其它高溴代PBDEs强,很容易在贝类和鱼体等海洋生物中累积[10 − 11]. 贻贝对多数污染物有高蓄积性和耐受性,常作为海洋污染的指示生物,以鉴别污染物的时空分布特征和毒性作用[12]. 研究表明,BDE-47可在多种海洋贝类中蓄积转化并引发毒性效应[11,13]. 由BDE-47引发的生态和健康风险受到广泛关注[14 − 15].
2-羟丙基-β-环糊精(2-hydroxypropy-β-cyclodextrin, HPCD)是一种β-环糊精(β-CD)衍生物,具有疏水性空腔和亲水外围表面,被广泛应用于食品保鲜、药物传递和环境污染治理等领域[16]. HPCD可以通过包合、吸附等方式,将污染物包裹在空腔内部,可以促进有机污染物在环境介质中的传递,促进生物降解;减少污染物与生物的直接作用或将生物体中结合态污染物解吸出来,降低蓄积和毒性作用[17]. 近年来,环糊精及其衍生物也被添加到阻燃剂中,以增强其性能同时减少阻燃剂组分向环境中释放,在污染物的减毒和消除方面发挥重要作用,BDE-47作为多种PBDEs代谢物,在实际环境中极有可能与HPCD共存[18 − 22].
基于以上研究背景,本实验以BDE-47污染风险严峻的紫贻贝为研究对象,比较HPCD加入后BDE-47在贻贝各组织中的蓄积和分布变化,并计算BDE-47在贻贝各组织中蓄积和消除的动力学参数,测定氧化胁迫指标和组织损伤情况,结合分子对接模拟,以揭示HPCD对BDE-47生物蓄积、消除和毒性的影响.
1. 材料与方法(Materials and methods)
1.1 实验材料
1.1.1 仪器与试剂
一台Agilent 7890B气相色谱仪,配备μECD检测器和DB-5MS色谱柱(15 m×0.25 mm×0.10 μm). 一台HS-
3345 旋转切片机,购自金华市华速科技有限公司. BDE-47标准品(纯度>98%,Wellington Laboratories)购自北京联众行贸易有限公司,BDE-47粉末(>95%)购自上海源叶生物科技有限公司. 2-羟丙基-β-环糊精(HPCD,>98%)购自北京沃凯生物科技有限公司. Captiva EMR-Lipid固相萃取柱(6 mL,600 mg)购自美国Agilent Technologies公司. 乙腈、水和二甲基亚砜(DMSO)等均为HPLC级别,购自美国Merck公司. 超氧化物歧化酶(SOD)和谷胱甘肽-S-转移酶(GST)活性、还原型谷胱甘肽(GSH)和丙二醛(MDA)含量测定试剂盒购自南京建成生物工程研究所. Bouin’s固定液购自北京兰博康斯科技有限公司. 其他试剂未作说明均为分析纯,购自国药集团化学试剂有限公司.1.1.2 供试生物
二龄紫贻贝采集自山东青岛胶州湾养殖场,体重(12.7±1.6)g,壳长(5.4 ± 0.62)cm. 实验开始前,在洁净过滤海水中驯养10 d以适应实验室条件. 实验用水温度(15.2 ± 1.9)℃,pH 7.98 ± 0.15,盐度37.9% ± 2.1%(M:M),光照/黑暗周期为14 h/10 h. 期间不间断曝气,每天定时投喂1次螺旋藻粉(0.06%体重比),暂养期间贻贝死亡率<1%.
1.2 实验方法
1.2.1 暴露实验与样本采集
暴露实验:称取BDE-47溶于DMSO中,配制100 μg·mL−1的BDE-47储备液,实验时用海水稀释至所需刻度. 暂养结束后选取健康且大小均一的紫贻贝,随机分到12只养殖箱中(90 cm×60 cm×30 cm,装有50 L溶液),每个养殖箱中60只. 12只养殖箱分为4组:海水对照组、溶剂对照组、BDE-47处理组(10 ng·mL−1)和BDE-47+HPCD处理组(10 ng·mL−1 BDE-47+60 ng·mL−1 HPCD,物质的量比约为1:2),每组设置3个重复. 实验在半静态条件下开展,每24 h更换等浓度暴露溶液,其它条件与暂养时一致.
消除实验:暴露实验后,将紫贻贝转移到洁净海水中净化10 d至无BDE-47检出,其中BDE-47+ HPCD处理组继续添加60 ng·mL−1的HPCD直至实验结束.
1.2.2 样本前处理与仪器分析
在暴露实验开始的第0、1、3、5、7、14、21、28 d随机取12—15只(每个养殖箱4—5只)贻贝,解剖分离消化盲囊、鳃、性腺、外套膜和闭壳肌等部位,合并同一养殖箱的组织样本,冻干后研磨成粉末用于BDE-47含量测定. BDE-47含量测定参考耿倩倩和Komolafe的方法[11,23],并根据实际情况优化,具体方法如下:准确称取(0.20 ± 0.01)g冻干粉,加入10 mL乙腈-水(80:20,V:V),1.0 g氯化钠并涡旋30 s,水浴条件下超声辅助萃取10 min,之后
4000 r·min−1离心5 min,移取5 mL上清液过Captiva EMR-Lipid小柱净化,收集萃取液,40 ℃氮气吹干,加入1 mL正己烷溶解,过0.22 μm滤膜后待测.仪器分析:气相色谱(GC)程序升温条件:100 ℃保持1 min,20 ℃·min−1升温至320 ℃保持5 min. 进样量:1 μL,进样方式:不分流. 外标法定量BDE-47含量以干重计(μg·g−1).
质量控制:同时做方法空白、空白加标和基质加标,以控制整个分析过程的准确度和精密度.
1.2.3 氧化损伤等生化指标测定
基于预实验测定结果,选取暴露28 d的6—10只贻贝,冰上快速解剖分离消化盲囊和性腺,以1:9(V:V)体积比加入遇冷生理盐水匀浆,4 ℃下
5000 r·min−1离心10 min,取上清液待测;GST活性测定采用比色法,以GSH浓度降低的方式反映;还原型GSH含量测定原理是GSH可与二硫代二硝基苯甲酸(DTNB)反应,生成一种黄色化合物,可在405 nm下进行比色定量测定;SOD活性测定以反应体系中SOD抑制率对应的酶量表示,CAT活性测定采用钼酸铵法,MDA含量测定使用TBA,具体测定方法参照检测试剂盒说明书操作. 参考文献[24]中方法,将所得指标进行均一化处理,绘制响应星状图,计算综合生物标志物响应(IBR)指数. 贻贝脂肪含量测定采用酸水解法(GB/T5009.6 —2016 食品安全国家标准食品中脂肪的测定).1.2.4 组织切片制备
选取暴露28 d的3—5只贻贝,解剖分离消化盲囊和性腺,置于Bouin’s固定液中固定48 h,用于制备组织病理切片. 经梯度乙醇溶液脱水,二甲苯、甲苯和石蜡透明处理,浸蜡和包埋后,将蜡块修整为1 cm3左右,在旋转切片机中切成厚度介于3—5 μm的切片,之后经过烤片、贴片和苏木精-伊红染色(H&E染色)处理后,置于光学显微镜下观察.
1.2.5 分子对接
分子对接用于模拟BDE-47与HPCD可能的相互作用模式. BDE-47和HPCD的3D结构用Chem Bio Office 构建,将BDE-47定义为配体,HPCD 作为受体,在MMFF94x力场下将配体和受体分子能量最小化. 对接时,配体分子构象设为柔性可旋转,迭代次数为
1000 次,梯度测试值为 0.01,根据对接能量(S值)选择最优得分构象.1.2.6 蓄积与消除动力学分析
采用Origin 2021软件中的一阶非线性蓄积-消除模型,对每个处理组贻贝组织在各取样时间点的BDE-47平均含量进行蓄积和消除动力学拟合[11]. 首先按照公式(1)计算消除动力学参数(Ke, d−1):
stringUtils.convertMath(!{formula.content}) (1) 式中,Ct是消除实验在时间t时贻贝组织中BDE-47含量(ng·g−1),Ct=0是消除实验开始时对应组织中BDE-47的含量(ng·g−1).
按照公式(2)的非线性回归,计算蓄积速率常数(Ka, mL·g−1·d−1):
stringUtils.convertMath(!{formula.content}) (2) 式中,Ct是蓄积实验在时间t时贻贝组织中BDE-47含量(ng·g−1),C是暴露溶液中BDE-47的浓度(ng·mL−1).
按照公式(3)计算消除半衰期(t1/2, d):
stringUtils.convertMath(!{formula.content}) (3) 动力学来源生物富集因子(BCFk, mL·g−1)按照公式(4)计算:
stringUtils.convertMath(!{formula.content}) (4) 式中,Ka和Ke值分别为计算所得蓄积和消除速率常数.
依据公式(5)计算观测所得生物富集系数(BCFo, mL·g−1):
stringUtils.convertMath(!{formula.content}) (5) 式中,Cm为蓄积实验结束时贻贝组织中BDE-47平均含量(ng·g−1),Cw为暴露溶液中BDE-47实测浓度(ng·mL−1).
1.3 数据分析
本研究中实验数据的统计使用SPSS 20.0软件,Duncan’s法进行多重比较,显著水平P = 0.05,以均值± SD值表示,使用Origin 2021软件绘图.
2. 结果与讨论(Results and discussion)
2.1 HPCD对BDE-47在贻贝中蓄积、分布和消除的影响
蓄积和代谢消除阶段,对照组和各处理组紫贻贝的生长状态良好,死亡率均<1%. 紫贻贝消化盲囊、性腺、鳃、闭壳肌、外套膜和整贝(软组织)中BDE-47含量-时间变化如图1所示,拟合所得蓄积和消除动力学参数列于表1.
 图 1 蓄积和消除过程中四溴联苯醚(BDE-47)在贻贝消化盲囊、性腺、鳃、闭壳肌、外套膜和整贝(软组织)中的含量变化Figure 1. Changes of tetrabromodiphenyl ether (BDE-47) content in digestive gland, gonad, gill, mantle, adductor muscle and whole mussel (soft tissue) of blue mussel during accumulation and elimination(n=3, dry weight, mean ± SD)(n=3,含量以干重计算,均值±SD)表 1 BDE-47处理组和BDE-47+HPCD处理组的紫贻贝组织中BDE-47的蓄积和消除动力学参数Table 1. The accumulation and elimination kinetic parameters of BDE-47 in the tissues of blue mussels treated with BDE-47 and BDE-47+HPCD
图 1 蓄积和消除过程中四溴联苯醚(BDE-47)在贻贝消化盲囊、性腺、鳃、闭壳肌、外套膜和整贝(软组织)中的含量变化Figure 1. Changes of tetrabromodiphenyl ether (BDE-47) content in digestive gland, gonad, gill, mantle, adductor muscle and whole mussel (soft tissue) of blue mussel during accumulation and elimination(n=3, dry weight, mean ± SD)(n=3,含量以干重计算,均值±SD)表 1 BDE-47处理组和BDE-47+HPCD处理组的紫贻贝组织中BDE-47的蓄积和消除动力学参数Table 1. The accumulation and elimination kinetic parameters of BDE-47 in the tissues of blue mussels treated with BDE-47 and BDE-47+HPCD组别Group 组织Tissue Ke/d−1 Ka/(mL·g−1·d−1) t1/2/d BCFk/(mL·g−1) BCFo/(mL·g−1) BDE-47 消化盲囊(digestive gland) 0.2447 720.6 2.83 2944.8 5725.5 性腺(gonad) 0.2595 580.1 2.67 2235.5 5032.2 鳃(gill) 0.5322 913.2 1.30 1715.9 3587.8 外套膜(mantle) 0.1932 257.9 3.59 1334.9 2898.5 闭壳肌(adductor muscle) 0.2363 284.0 2.93 1201.9 2365.2 整贝(whole mussel) 0.2152 428.9 3.22 1993.0 3983.4 BDE-47+HPCD 消化盲囊(digestive gland) 0.2875 579.4 2.41 2015.3 4214.8 性腺(gonad) 0.2521 384.0 2.75 1523.2 3693.9 鳃(gill) 0.6622 840.5 1.05 1269.3 2646.8 外套膜(mantle) 0.3718 423.3 1.86 1138.5 2329.7 闭壳肌(adductor muscle) 0.2171 203.4 3.19 936.9 2194.7 整贝(whole mussel) 0.4892 702.2 1.42 1435.4 3092.7 | Show Table DownLoad:
CSV
DownLoad:
CSV
(1)BDE-47蓄积和分布.
暴露开始后1 d,紫贻贝各部位均可检测到BDE-47,随暴露时间延长,BDE-47含量均逐渐增大,至第28天时达到最大值;BDE-47处理组与BDE-47+HPCD处理组蓄积趋势一致:在暴露阶段BDE-47含量持续增加,至第28天仍未观察到平台期,说明紫贻贝对BDE-47的蓄积能力较强;各取样时间点的紫贻贝组织中BDE-47含量顺序大致为:消化盲囊>性腺>鳃>闭壳肌和外套膜,消化盲囊是BDE-47蓄积的靶组织.
在蓄积开始阶段(1—5 d),HPCD对紫贻贝中BDE-47的蓄积量无显著影响,各处理组的BDE-47含量接近;随暴露时间延长,BDE-47+HPCD处理组的贻贝组织中BDE-47含量总体上低于BDE-47处理组,这一现象在消化盲囊和性腺中更为明显,HPCD降低了BDE-47在紫贻贝各组织中的蓄积浓度.
(2)BDE-47消除特征
在消除阶段的前期,BDE-47含量降低较为明显,后期消除速率有不同程度的减缓. 经过10 d的消除,各处理组紫贻贝组织中仍有一定比例的BDE-47残留,BDE-47处理组残留量约为蓄积终点含量的20.4%—50.7%,BDE-47+HPCD处理组为9.0%—20.9%;BDE-47残留量顺序:BDE-47处理组,性腺>鳃>消化盲囊>闭壳肌>外套膜;BDE-47+HPCD处理组,性腺>消化盲囊>外套膜>鳃>闭壳肌. 紫贻贝对BDE-47的消除能力较差,BDE-47在紫贻贝中存在较高的残留风险. 在消除阶段几乎所有时间点的含量均是BDE-47处理组>BDE-47+HPCD处理组,HPCD促进了紫贻贝对BDE-47的消除. 主要表现为HPCD加入后,BDE-47的消除速率Ke值增大,蓄积速率Ka值和富集系数(BCFk和BCF0)降低,并且具有更短的消除半衰期(表1). 紫贻贝整贝(软组织)中BDE-47的蓄积和消除的趋势与各组织类似.
BDE-47在紫贻贝中存在组织特异性蓄积和消除. BDE-47脂溶性较强,消化盲囊和性腺脂肪含量高于其它组织(消化盲囊13.4%,性腺13.7%,其它组织均<8%,以干重计),消化盲囊也是贝类代谢的重要部位,BDE-47经鳃等组织吸收后倾向于在这两种组织中蓄积,其它有机污染物在贻贝中蓄积和分布的组织差异也被证实[11,26]. BDE-47在贻贝性腺中的高浓度蓄积或可引发贝类繁殖异常. 鳃是BDE-47吸收和消除的重要部位,这与鳃丝有较大的接触表面积和丰富的血管有关. 研究表明,γ-CD可通过与全氟烷基辛酸(PFOA)形成包合物,逆转PFOA与血清蛋白(HSA)的结合,从而降低蓄积[27];而加入β-CD或者氨基键合的β-CD也有助于将全氟(2-甲基-3-氧杂己酸,GenX)从HAS-GenX复合体中提取出来,从而缓解污染[28]. 与上述环糊精相似,HPCD也具有疏水性内腔和亲水外壁,容易和疏水性的污染物通过非共价相互作用形成包合物,改变赋存形态,降低生物有效性从而降低蓄积;HPCD也可能逆转贻贝中BDE-47的结合,促进其代谢消除[18 − 19]. 模拟生物膜吸收研究表明,HPCD可降低多种有机污染物的表观渗透率从而降低污染物的吸收量[29],因此本研究中HPCD存在下降低BDE-47蓄积和促进消除作用可能是以上多因素影响的结果.
2.2 HPCD对BDE-47毒性效应的影响
2.2.1 HPCD对BDE-47所致氧化损伤影响
研究表明,BDE-47暴露可在许多无脊椎动物中引发过氧化应激反应,并诱导机体的抗氧化防御功能,保护生物体免受氧化损伤[25,29 − 30]. 本研究中,蓄积28 d的消化盲囊和性腺中抗氧化酶活性(GST、SOD和CAT)脂质过氧化生物标记物(MDA)和非酶抗氧化剂(GSH)均受到BDE-47和BDE-47+HPCD暴露的影响,相关结果列于图2. 与对照组相比,BDE-47和BDE-47+HPCD显著(P<0.05)抑制消化盲囊和性腺GST活性,并且BDE-47处理组抑制作用较大(图2A);GSH含量变化趋势与GST一致(图2B). BDE-47和BDE-47+HPCD暴露处理的SOD(图2C)和CAT(图2D)酶活性,MDA(图2E)含量均显著(P<0.05)高于对照组,并且前者MDA含量的诱导升高作用高于后者.
 图 2 紫贻贝消化盲囊和性腺中谷胱甘肽-S-转移酶活性(GST,A),还原型谷胱甘肽含量(GSH,B),超氧化物歧化酶(SOD,C)和过氧化氢酶活性(CAT,D),丙二醛含量变化(MDA,E),综合生物标志物星图(F-消化盲囊,G-性腺)Figure 2. Glutathione S-transferase activity (GST, A), glutathione content (GSH, B), superoxide dismutase (SOD, C) and catalase activity (CAT, D), malondialdehyde content change (MDA, E), integrated biomarker star chart (F-digestive gland, G-gonad) in digestive gland and gonad of blue mussel(28 d, n=3, mean ± SD, P=0.05)(28 d,n=3,均值±SD,P=0.05)
图 2 紫贻贝消化盲囊和性腺中谷胱甘肽-S-转移酶活性(GST,A),还原型谷胱甘肽含量(GSH,B),超氧化物歧化酶(SOD,C)和过氧化氢酶活性(CAT,D),丙二醛含量变化(MDA,E),综合生物标志物星图(F-消化盲囊,G-性腺)Figure 2. Glutathione S-transferase activity (GST, A), glutathione content (GSH, B), superoxide dismutase (SOD, C) and catalase activity (CAT, D), malondialdehyde content change (MDA, E), integrated biomarker star chart (F-digestive gland, G-gonad) in digestive gland and gonad of blue mussel(28 d, n=3, mean ± SD, P=0.05)(28 d,n=3,均值±SD,P=0.05)GST是一种关键的Ⅱ相解毒酶,GSH是一种重要的非酶抗氧化剂. GST催化GSH与许多亲电化学物质结合并生成水溶性物质,在外源化学物质的抗氧化防御中发挥重要作用. GST活性的降低表明生物体无法适应BDE-47暴露处理引起的过氧化胁迫,这与之前关于PBDEs等污染物对贻贝暴露胁迫的研究一致[31]. 与BDE-47处理组相比,BDE-47+HPCD暴露处理组GST活性和GSH含量降低幅度较小,说明后者氧化胁迫的程度更低. SOD是一种重要的抗氧化酶,因为它能将超氧自由基(O2−)分解为H2O2,以消除活性氧(ROS),保护机体免受氧化损伤. CAT是消除H2O2的酶,与SOD一起协同作用保护生物体维持稳态平衡,具有抗炎作用[32]. SOD和CAT活性的显著升高说明BDE-47诱导O2−生成造成氧化胁迫;MDA是生物体内膜脂过氧化的产物,MDA含量升高意味着O2−并没有被彻底清除,造成ROS的积累[24,33]. BDE-47+HPCD处理组MDA含量显著低于BDE-47处理组,表明HPCD减轻了BDE-47诱导的氧化胁迫作用. SOD、CAT、MDA测定结果与GST、GSH测定结果一致.
图2F,2G所示为综合生物标志物星图,各指标中以SOD的响应值最高,GST、GSH和MDA的响应值相近,CAT响应值最低. IBR值可以综合多种生物标志物对污染胁迫的反应,用于评估污染物毒性效应. IBR值越高,毒性反应越大[34]. 消化盲囊BDE-47和BDE-47+HPCD处理组的IBR值(图2F、G中相邻生物标志物的辐射线围成的星状图面积之和)分别为3.40和1.47,性腺中分别为1.26和0.82. 说明BDE-47处理组的各生物标志物指标对暴露胁迫的反应更高. 氧化应激各项指标和IBR值结果说明HPCD大幅度降低了BDE-47诱导的氧化胁迫和毒性作用.
2.2.2 HPCD对BDE-47所致消化盲囊和性腺病理变化的影响
(1)消化盲囊
对照组(图3A)消化盲囊中可见形态正常的消化管(dt),内含管腔(l)和分泌物(sp),外侧包围着单层或连续的消化细胞和嗜碱性细胞,内壁间质组织呈现“星形”结构,周围连接组织(ct)正常,未发现明显的萎缩或者坏死等结构损伤;BDE-47处理组(图3B)消化盲囊中观察到明显的消化管萎缩(at),组织脱落(ex),血细胞浸润(hi)和连接组织纤维化(fi)等损伤,内壁间质组织“星形”结构不明显. BDE-47+HPCD处理组(图3C)消化盲囊表现为消化管萎缩(at),组织脱落(ex)连接组织纤维化(fi),内壁间质组织“星形”结构同样不明显,组织损伤与BDE-47处理组无明显差别.
 图 3 紫贻贝消化盲囊和性腺的组织病理切片照片(28 d,H&E染色)Figure 3. Histopathological pictures of the digestive gland and gonad of blue mussel (28 d, H&E staining)
图 3 紫贻贝消化盲囊和性腺的组织病理切片照片(28 d,H&E染色)Figure 3. Histopathological pictures of the digestive gland and gonad of blue mussel (28 d, H&E staining)(2)性腺
对照组(图3D)雄性紫贻贝性腺处于生精中期,可见发育正常的精巢组织,大量初级次级精母细胞(*)呈辐射状排列,滤泡边界清晰可见;BDE-47处理组(图3E)精母细胞辐射状排列不规则,且精母细胞数量明显少于对照组,滤泡边界模糊;BDE-47+HPCD处理组(图3F)精巢组织的滤泡边缘模糊增厚,精母细胞数量和排列和对照组相比变化不大,精巢损伤低于BDE-47处理组.
对照组(图3G)可见发育完全的紫贻贝卵巢,表现为结构正常的梨形、圆形或卵圆形的卵细胞(1),大小均一,卵细胞内含细胞核(2)与核仁(3),细胞核比例适中,与核质界限分明,核仁清晰可见,着色较深;卵细胞外有滤泡壁(4)包围,滤泡间隙较小. BDE-47处理组(H)卵细胞数量明显减少,细胞核增大,核仁分散不清晰,且滤泡壁模糊增厚(5),相邻滤泡间隙变大(6). BDE-47+HPCD处理组(图3I)卵细胞数量变化不显著,但也表现为滤泡壁模糊增厚(7)和滤泡间隙增大(8)的现象,损伤程度低于BDE-47暴露组.
研究表明,PBDEs可诱导双壳贝类的炎症反应并导致消化管等的损伤[15, 35]. 本研究中消化管萎缩表现为管腔增大或小管厚度增加,这是由炎症反应造成的,消化管内腔组织脱落是由消化细胞坏死引起的,血细胞浸润是由于管腔细胞破损,血细胞进入管腔引发. HPCD对BDE-47蓄积的消化盲囊组织损伤影响较小,这可能是由于两个处理组的消化盲囊中均蓄积了较高浓度的BDE-47导致的. 基于脊椎动物的毒性实验表明,BDE-47具有生殖发育毒性和遗传毒性[36]. 本研究中各处理组紫贻贝性腺均有不同程度的损伤,比如生殖细胞数量降低等,说明BDE-47可能影响其生殖功能,由BDE-47引发的海洋无脊椎动物尤其是双壳贝类的生殖功能异常也应当引起足够重视. 组织病理研究结果表明HPCD一定程度上减轻了对紫贻贝消化盲囊和性腺的组织损伤.
2.2.3 分子对接
分子对接可直观展示主客体化合物之间的相互作用方式,优选后的包合物构型列于图4. 分子对接模拟结果表明,BDE-47分子部分进入HPCD空腔,分子在空腔内部折叠,位于空腔居中位置. 其中BDE-47的部分苯环,醚键和溴原子靠近HPCD小口端,部分基团暴露在空腔外部. 分子对接模拟计算BDE-47和HPCD的结合能为−4.16 cal·mol−1,说明BDE-47和HPCD可以形成较为稳定的包合物. 基于分子对接的结果,推测HPCD对BDE-47蓄积、分布的影响和毒性降低的作用可能是由于HPCD对游离态BDE-47(暴露溶液中)的屏蔽作用,或对结合态BDE-47(贻贝组织中)的解吸附作用,降低BDE-47的蓄积并加速消除,继而减缓其毒性作用[19].
 图 4 分子对接模拟的BDE-47与HPCD相互作用空间构型Figure 4. Optimized structures of BDE-47 included into HPCD (a, b-side view; c, d-top view)(a,b-侧视图,c,d-俯视图)
图 4 分子对接模拟的BDE-47与HPCD相互作用空间构型Figure 4. Optimized structures of BDE-47 included into HPCD (a, b-side view; c, d-top view)(a,b-侧视图,c,d-俯视图)3. 结论(Conclusion)
(1)BDE-47在紫贻贝中的蓄积具有组织特异性;HPCD显著降低BDE-47在紫贻贝各组织中的蓄积浓度,加快BDE-47的清除:加入HPCD后,BDE-47蓄积速率常数(Ka)、消除半衰期(t1/2)和生物富集因子(BCFk,BCFo)减小,消除动力学参数(Ke)增大.
(2)HPCD减轻了BDE-47对紫贻贝消化盲囊和性腺的氧化胁迫和组织损伤;HPCD降低BDE-47在紫贻贝中的残留风险的同时也减轻了BDE-47对紫贻贝的毒性.
(3)BDE-47蓄积、分布和消除动态,以及氧化损伤的结果表明贻贝消化盲囊和性腺组织损伤减轻与HPCD降低BDE-47的蓄积量并减轻氧化胁迫有关.
(4)分子对接结果表明HPCD可能通过对BDE-47的屏蔽或者解吸附作用降低BDE-47在贻贝中的蓄积,继而减缓其毒性作用. 研究结果为HPCD在减少海洋生物污染物蓄积和促进消除方面的应用提供参考.
-
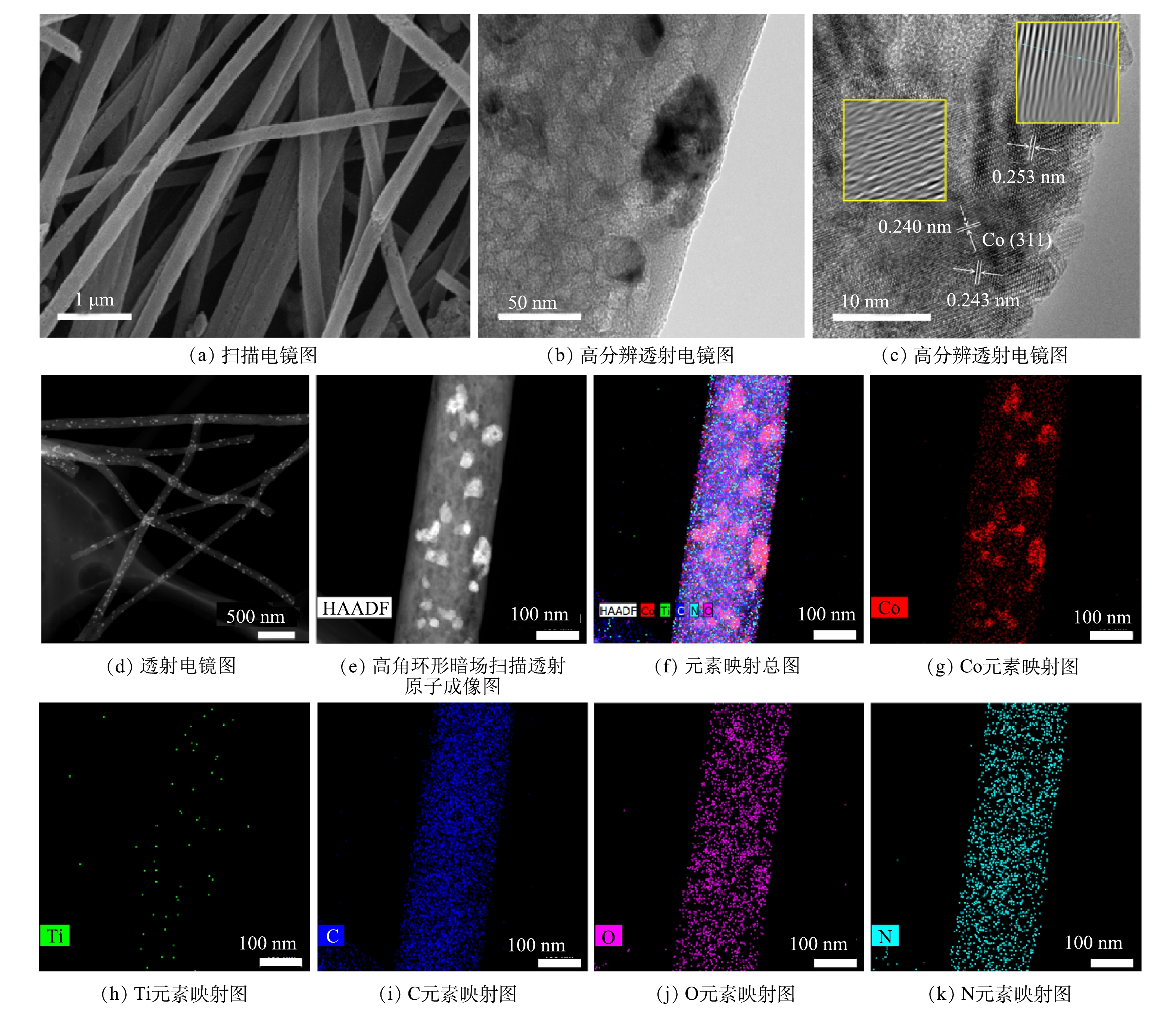
图 2 Co/TiO2@CNFs的形貌及表面元素分布分析
Figure 2. Morphology and surface element distribution analysis of Co/TiO2@CNFs
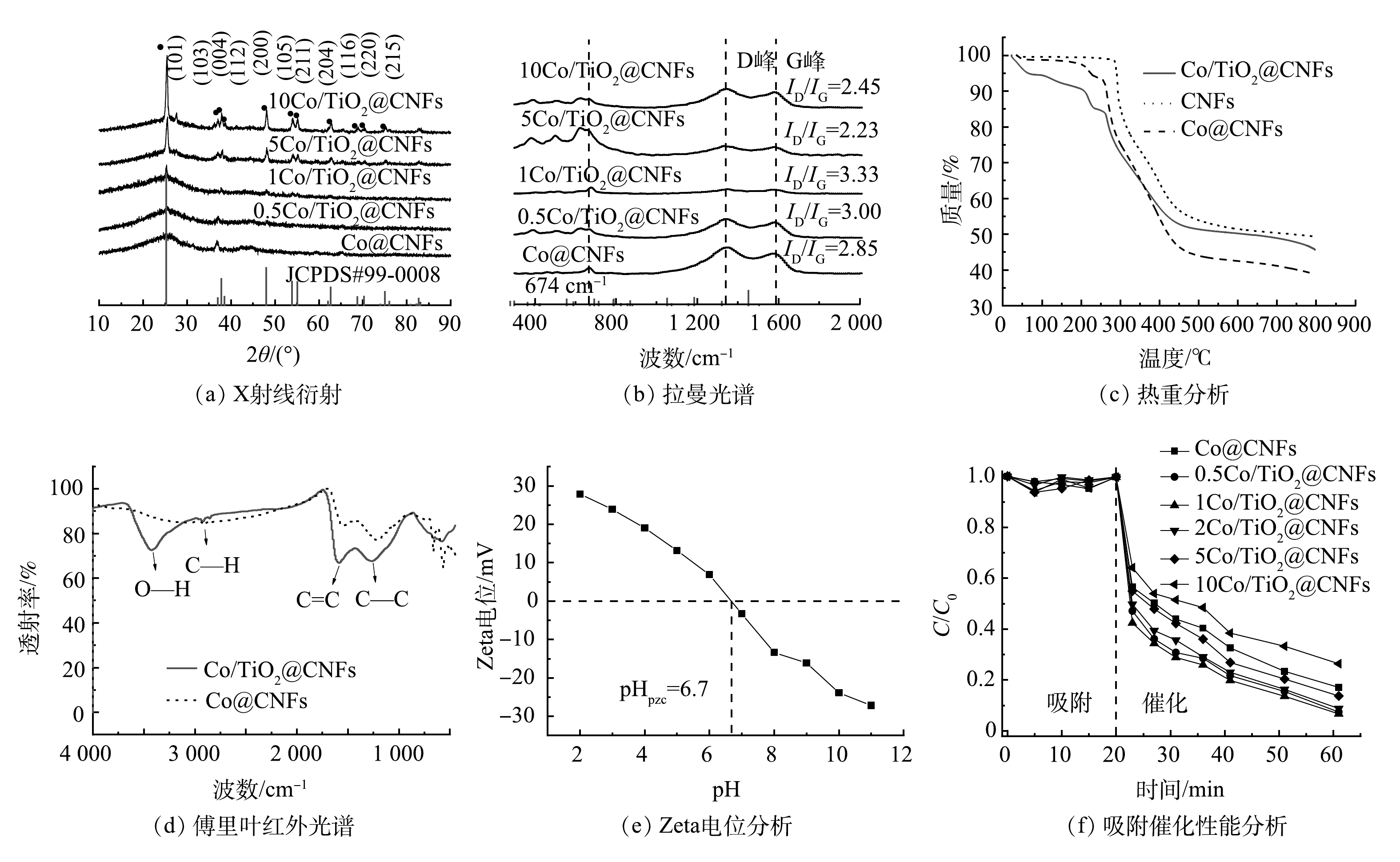
图 3 Co@CNFs与X Co/TiO2@CNFs的材料性能分析
Figure 3. Material properties analysis of Co@CNFs and X Co/TiO2@CNFs
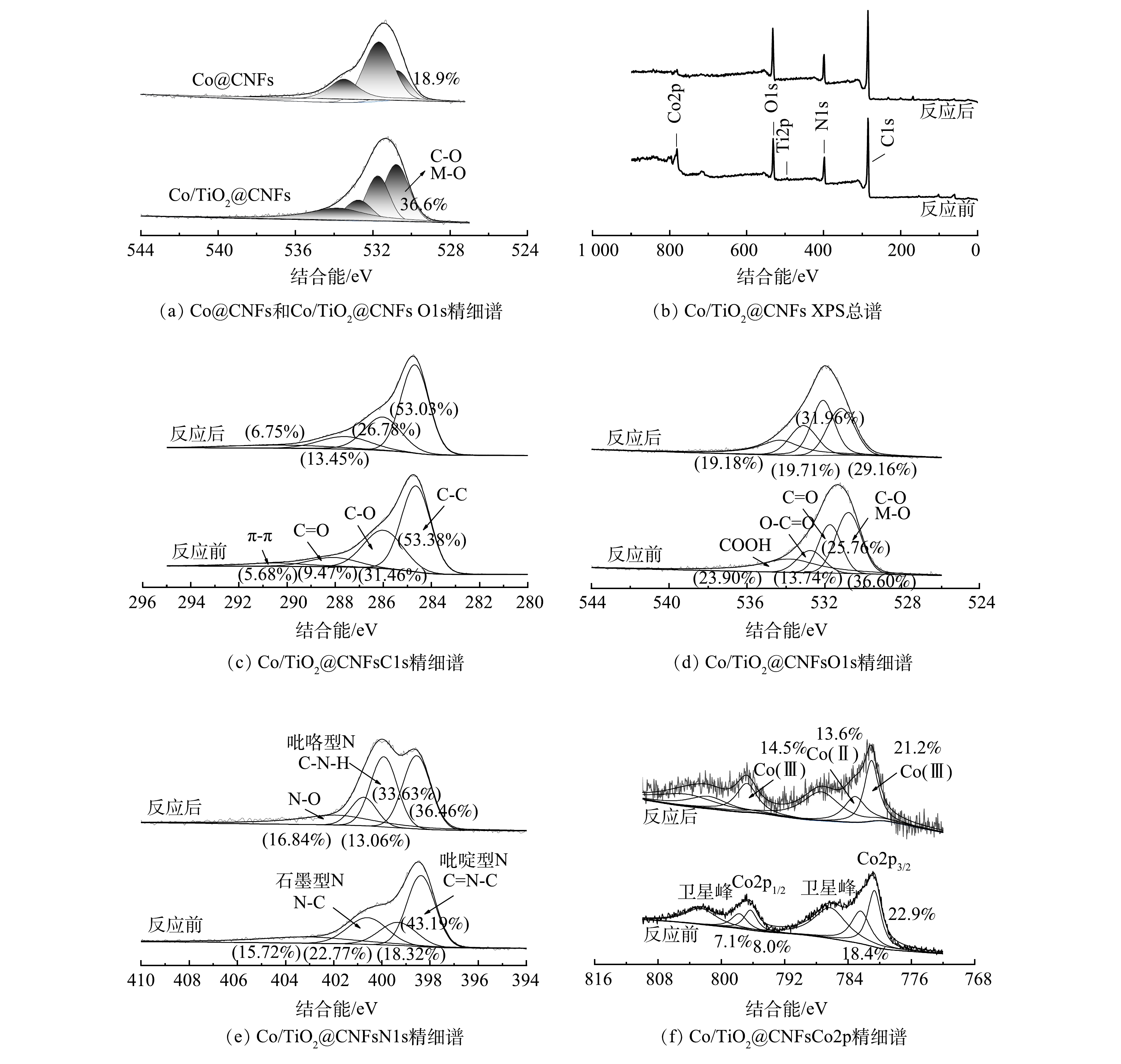
图 4 Co@CNFs和Co/TiO2@CNFs XPS O1s精细谱对比及Co/TiO2@CNFs反应前后XPS分析
Figure 4. XPS O1s spectra of Co@CNFs and Co/TiO2@CNFs, XPS spectra of Co/TiO2@CNFs before and after reaction
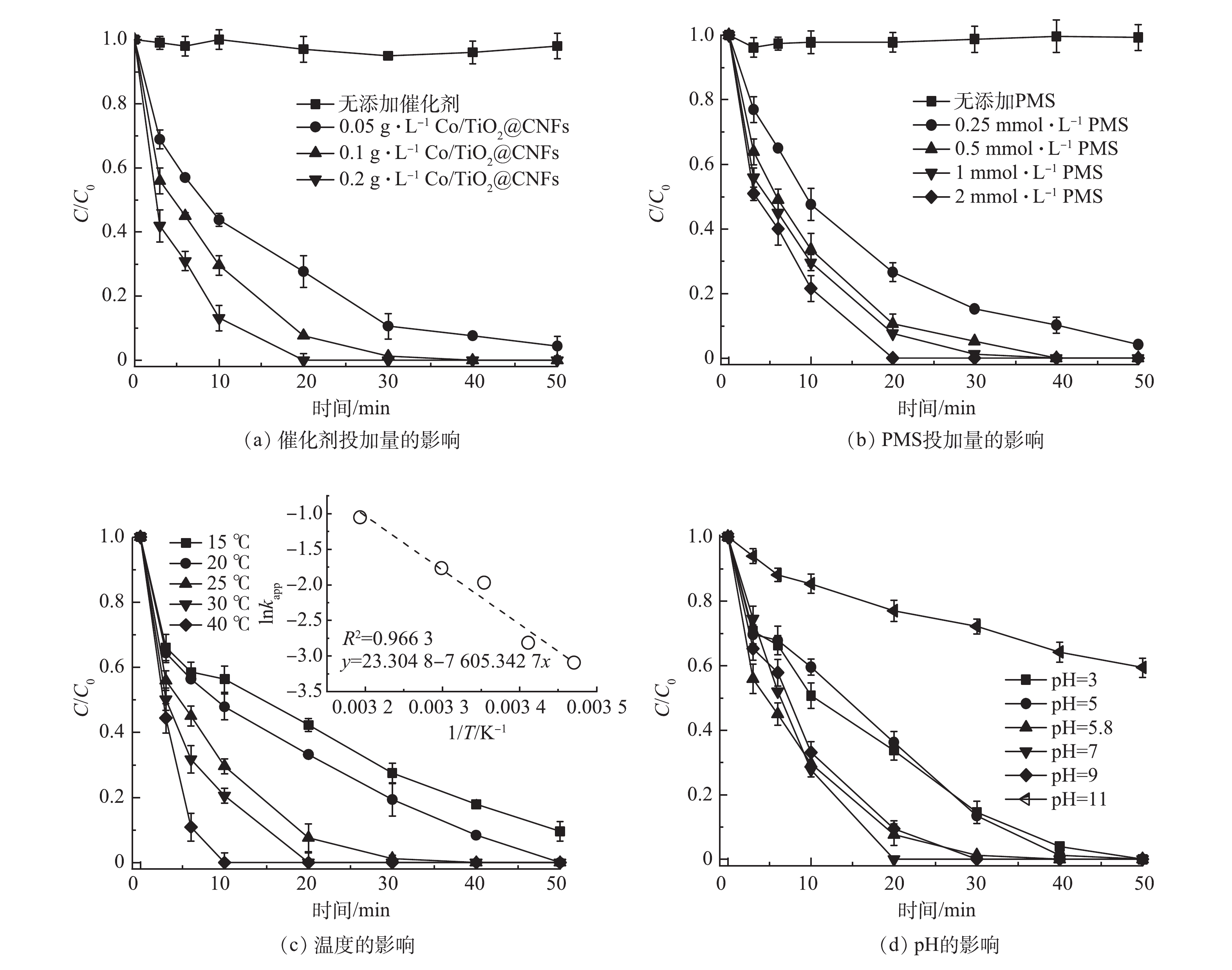
图 5 Co/TiO2@CNFs活化PMS降解SMX的影响因素分析
Figure 5. Effects of various affecting factors on SMX degradation by Co/TiO2@CNFs activated PMS
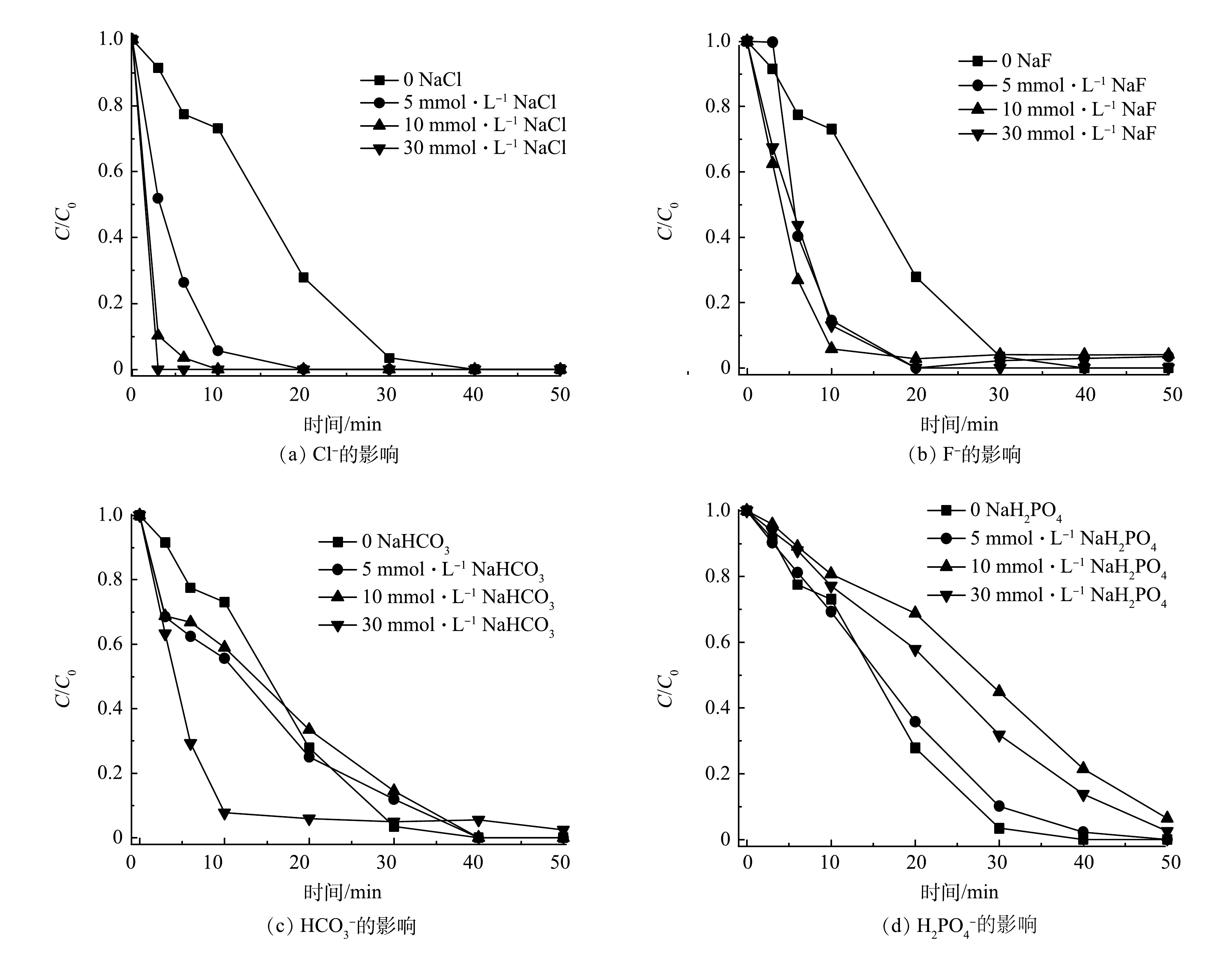
图 6 共存离子对Co/TiO2@CNFs活化PMS降解SMX的影响
Figure 6. Effect of coexisting ions on degradation of SMX by Co/TiO2@CNFs activated PMS
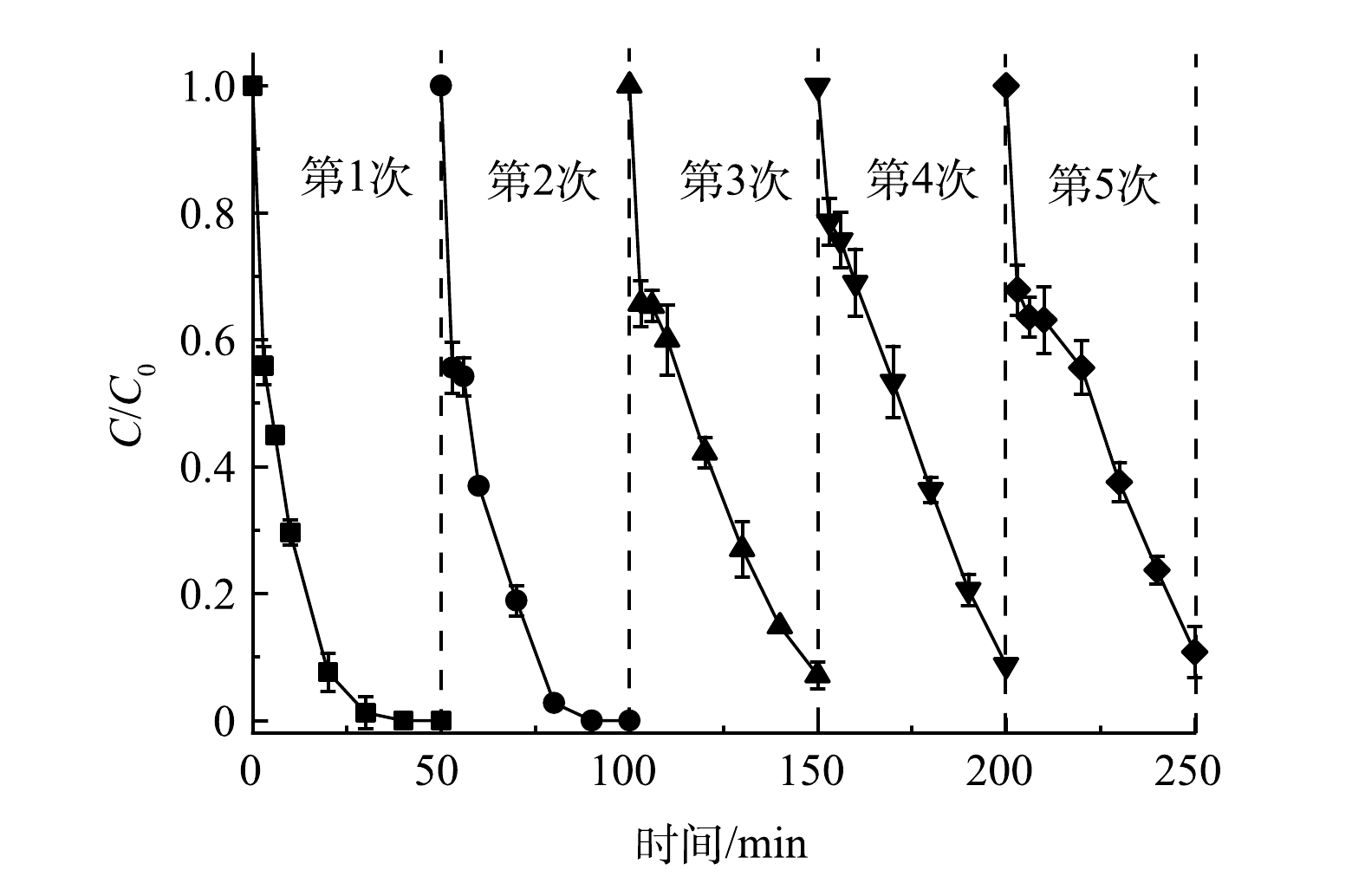
图 7 Co/TiO2@CNFs在活化PMS体系中的重复利用性
Figure 7. Reusability of Co/TiO2@CNFs in Co/TiO2@CNFs-activated PMS system
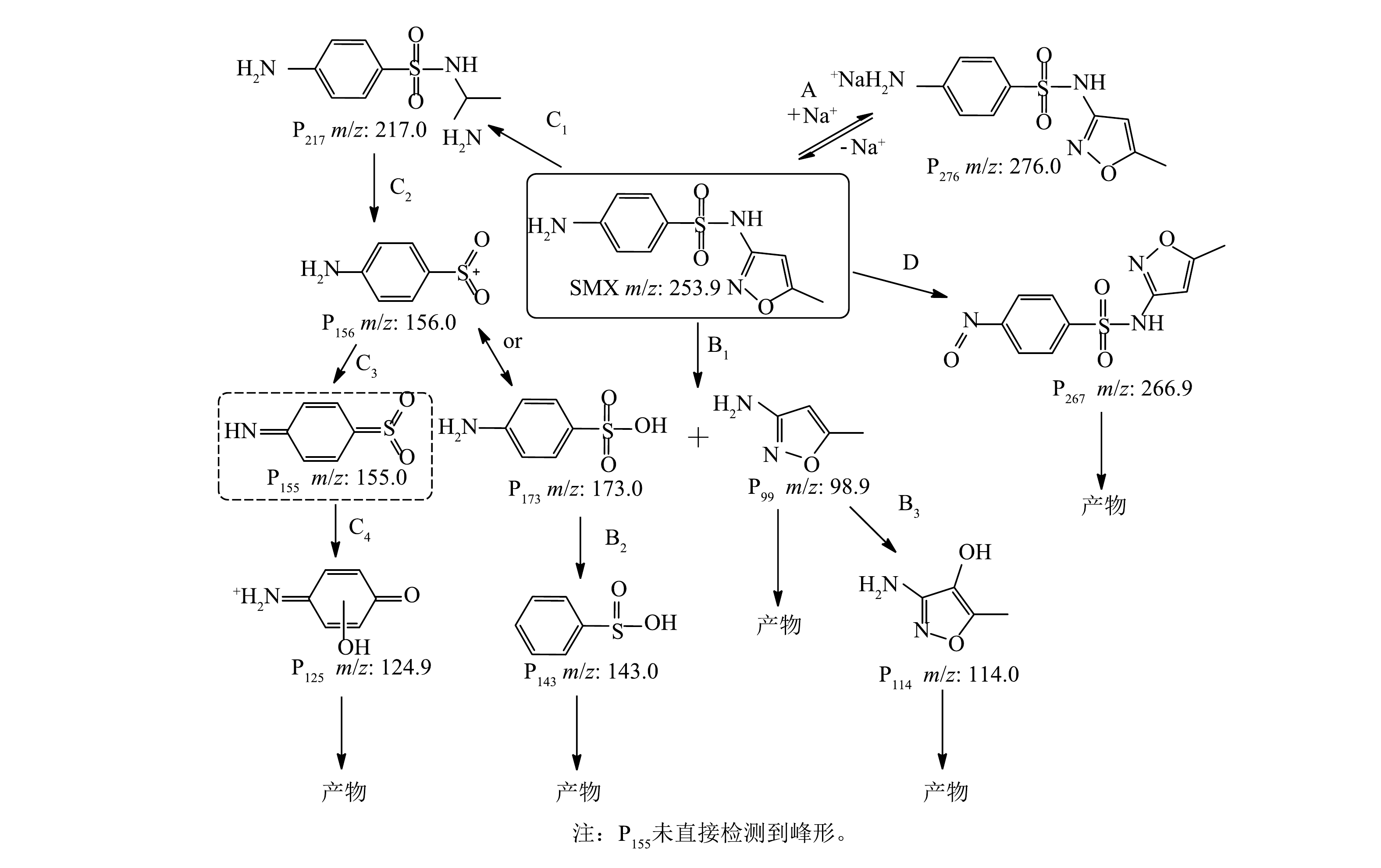
图 8 Co/TiO2@CNFs活化PMS降解SMX的路径分析
Figure 8. Proposed transformation pathways for SMX degradation in Co/TiO2@CNFs-activated PMS system
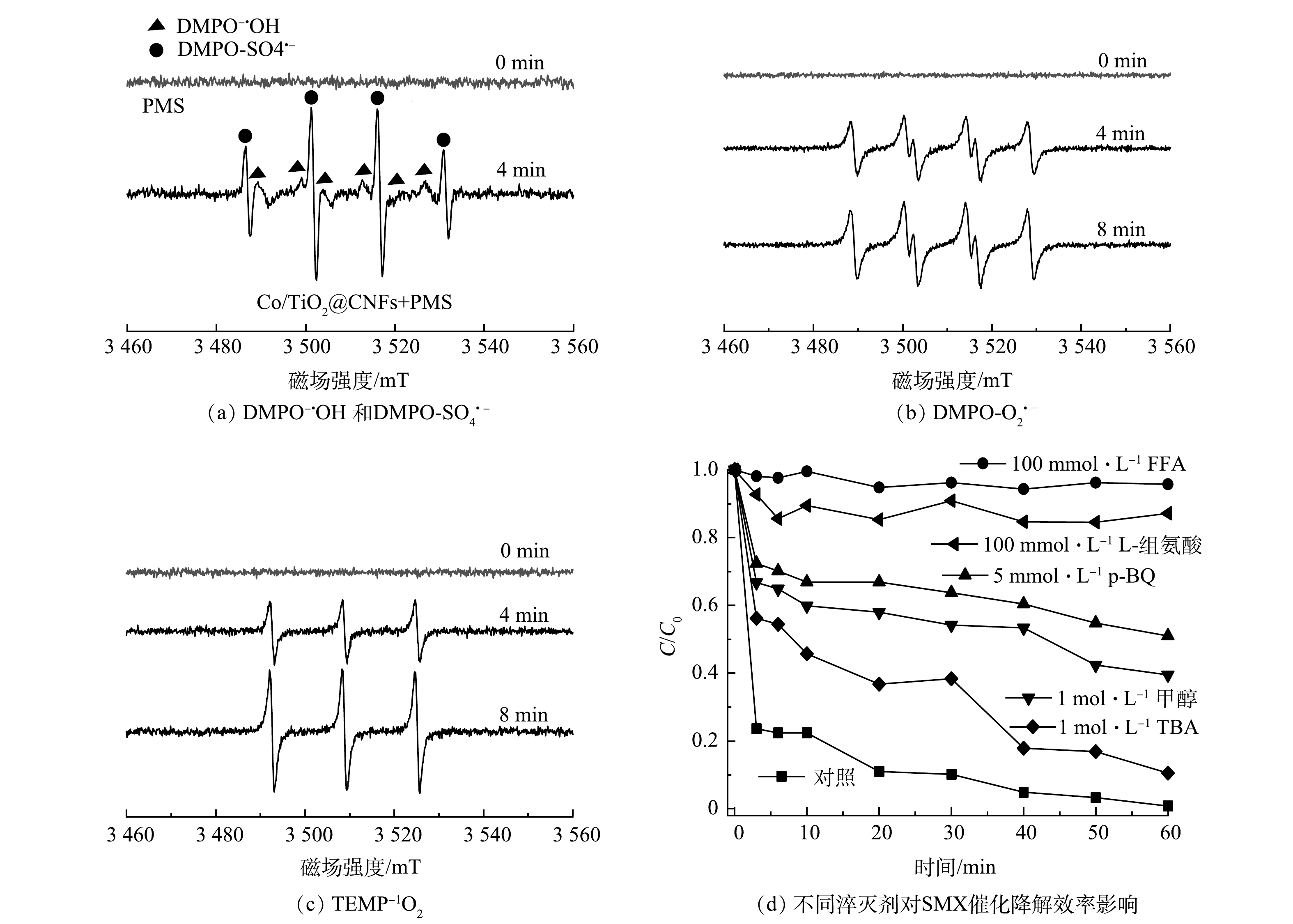
图 9 Co/TiO2@CNFs的电子自旋共振、淬灭实验
Figure 9. ESR spectra and quenching experiments of Co/TiO2@CNFs
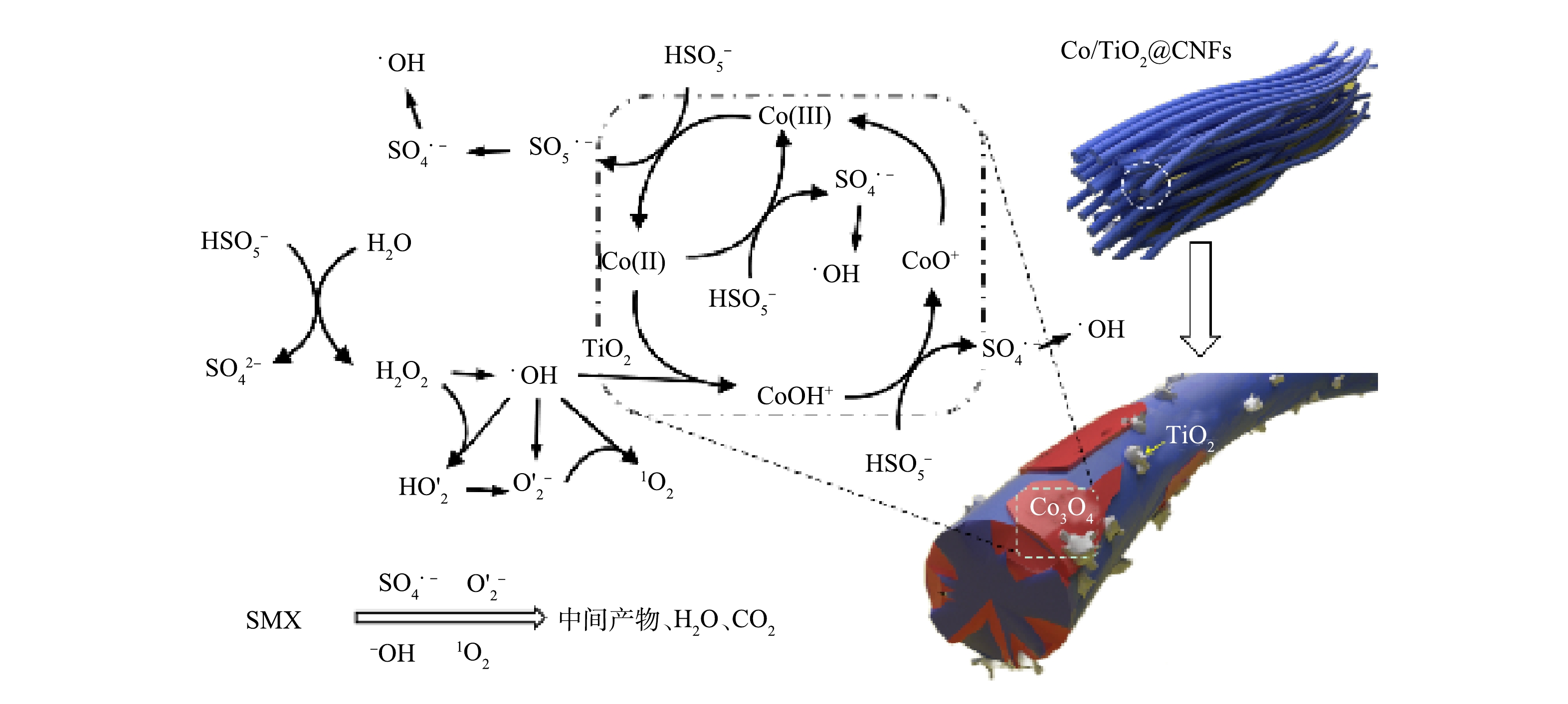
图 10 Co/TiO2@CNFs活化PMS降解SMX的机理分析
Figure 10. Reaction mechanism for the degradation of SMX in the Co/TiO2@CNFs-activated PMS system
表 1 Co@CNFs和X Co/TiO2@CNFs的N2吸脱附测试结果
Table 1. Results of N2 adsorption/desorption analysis of the Co@CNFs and X Co/TiO2@CNFs
材料 比表面积/(m2·g−1) 总孔容/(cm3·g−1) t-图法微孔/(cm3·g−1) 介孔/(cm3·g−1) 孔径/nm Co@CNFs 64.31 0.073 0.008 0.065 3.94 0.5Co/TiO2@CNFs 7.60 0.017 0.002 0.015 3.94 1Co/TiO2@CNFs 97.15 0.092 0.018 0.074 3.93 5Co/TiO2@CNFs 13.84 0.041 0.002 0.039 1.87 10Co/TiO2@CNFs 27.90 0.085 0.004 0.080 4.31
下载: 导出CSV
-
[1] HOU L, ZHANG H, WANG L, et al. Removal of sulfamethoxazole from aqueous solution by sono-ozonation in the presence of a magnetic catalyst[J]. Separation and Purification Technology, 2013, 117: 46-52. doi: 10.1016/j.seppur.2013.05.014 [2] YU X, SUI Q, LYU S, et al. Municipal solid waste landfills: An underestimated source of pharmaceutical and personal care products in the water environment[J]. Environment Science Technology, 2020, 54(16): 9757-9768. doi: 10.1021/acs.est.0c00565 [3] FEKADU S, ALEMAYEHU E, DEWIL R, et al. Pharmaceuticals in freshwater aquatic environments: A comparison of the African and European challenge[J]. Science of the Total Environment, 2019, 654: 324-337. doi: 10.1016/j.scitotenv.2018.11.072 [4] HIRSCH R, TERNES T, HABERER K, et al. Occurrence of antibiotics in the aquatic environment[J]. Science of the Total Environment, 1999, 225(1-2): 109-118. doi: 10.1016/S0048-9697(98)00337-4 [5] LI Y, SALLACH J B, ZHANG W, et al. Insight into the distribution of pharmaceuticals in soil-water-plant systems[J]. Water Research, 2019, 152: 38-46. doi: 10.1016/j.watres.2018.12.039 [6] SHANABLEH A, BHATTACHARJEE S, ALANI S, et al. Assessment of sulfamethoxazole removal by nanoscale zerovalent iron[J]. Science of the Total Environment, 2021, 761: 143307. doi: 10.1016/j.scitotenv.2020.143307 [7] TROVO A G, NOGUEIRA R F, AGUERA A, et al. Degradation of sulfamethoxazole in water by solar photo-Fenton. Chemical and toxicological evaluation[J]. Water Research, 2009, 43(16): 3922-3931. doi: 10.1016/j.watres.2009.04.006 [8] LIU F, ZHOU H, PAN Z, et al. Degradation of sulfamethoxazole by cobalt-nickel powder composite catalyst coupled with peroxymonosulfate: Performance, degradation pathways and mechanistic consideration[J]. Journal of Hazardous Materials, 2020, 400: 123322. doi: 10.1016/j.jhazmat.2020.123322 [9] WALTER M V, VENNES J W. Occurrence of multiple-antibiotic-resistant enteric bacteria in domestic sewage and oxidation lagoons[J]. Applied Environmental Microbiology, 1985, 50(4): 930-933. doi: 10.1128/aem.50.4.930-933.1985 [10] 许晟硕, 钱征, 王龄侦, 等. 氮掺杂碳催化剂活化过一硫酸盐的活性位点分析及其对双酚A的降解机制[J]. 环境工程学报, 2022,16(2): 452-461. [11] ZENG H, DENG L, ZHANG H, et al. Development of oxygen vacancies enriched CoAl hydroxide@hydroxysulfide hollow flowers for peroxymonosulfate activation: A highly efficient singlet oxygen-dominated oxidation process for sulfamethoxazole degradation[J]. Journal of Hazardous Materials, 2020, 400: 123297. doi: 10.1016/j.jhazmat.2020.123297 [12] ANDREOZZI R, CAPRIO V, INSOLA A, et al. Advanced oxidation processes (AOP) for water purification and recovery[J]. Catalysis Today, 1999, 53(1): 51-59. doi: 10.1016/S0920-5861(99)00102-9 [13] ZHANG H, LI G, DENG L, et al. Heterogeneous activation of hydrogen peroxide by cysteine intercalated layered double hydroxide for degradation of organic pollutants: Performance and mechanism[J]. Journal of Colloid and Interface Science, 2019, 543: 183-191. doi: 10.1016/j.jcis.2019.02.059 [14] ZENG H, ZHANG W, DENG L, et al. Degradation of dyes by peroxymonosulfate activated by ternary CoFeNi-layered double hydroxide: Catalytic performance, mechanism and kinetic modeling[J]. Journal of Colloid and Interface Science, 2018, 515: 92-100. doi: 10.1016/j.jcis.2018.01.016 [15] DUAN X, YANG S, WACŁAWEK S, et al. Limitations and prospects of sulfate-radical based advanced oxidation processes[J]. Journal of Environmental Chemical Engineering, 2020, 8(4). [16] CAI T, LIU Y, WANG L, et al. Activation of persulfate by photoexcited dye for antibiotic degradation: Radical and nonradical reactions[J]. Chemical Engineering Journal, 2019: 375. [17] HU P, LONG M. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications[J]. Applied Catalysis B:Environmental, 2016, 181: 103-117. doi: 10.1016/j.apcatb.2015.07.024 [18] BAO Y, TIAN M, LUA S K, et al. Spatial confinement of cobalt crystals in carbon nanofibers with oxygen vacancies as a high-efficiency catalyst for organics degradation[J]. Chemosphere, 2020, 245: 125407. doi: 10.1016/j.chemosphere.2019.125407 [19] INAGAKI M, YANG Y, KANG F. Carbon nanofibers prepared via electrospinning[J]. Advanced Materials, 2012, 24(19): 2547-2566. doi: 10.1002/adma.201104940 [20] ZHU Z, JI C, ZHONG L, et al. Magnetic Fe-Co crystal doped hierarchical porous carbon fibers for removal of organic pollutants[J]. Journal of Materials Chemistry A, 2017, 5(34): 18071-18080. doi: 10.1039/C7TA03990E [21] LIN K A, YANG M T, LIN J T, et al. Cobalt ferrite nanoparticles supported on electrospun carbon fiber as a magnetic heterogeneous catalyst for activating peroxymonosulfate[J]. Chemosphere, 2018, 208: 502-511. doi: 10.1016/j.chemosphere.2018.05.127 [22] ZHANG Y, ZHANG B T, TENG Y, et al. Carbon nanofibers supported Co/Ag bimetallic nanoparticles for heterogeneous activation of peroxymonosulfate and efficient oxidation of amoxicillin[J]. Journal of Hazardous Materials, 2020, 400: 123290. doi: 10.1016/j.jhazmat.2020.123290 [23] YANG Z, LI Y, ZHANG X, et al. Sludge activated carbon-based CoFe2O4-SAC nanocomposites used as heterogeneous catalysts for degrading antibiotic norfloxacin through activating peroxymonosulfate[J]. Chemical Engineering Journal, 2020, 384: 123319. doi: 10.1016/j.cej.2019.123319 [24] CAI C, ZHANG H, ZHONG X, et al. Electrochemical enhanced heterogeneous activation of peroxydisulfate by Fe–Co/SBA-15 catalyst for the degradation of Orange II in water[J]. Water Research, 2014, 66: 473-485. doi: 10.1016/j.watres.2014.08.039 [25] CHLEBDA D K, JĘDRZEJCZYK R J, JODŁOWSKI P J, et al. Surface structure of cobalt, palladium, and mixed oxide-based catalysts and their activity in methane combustion studied by means of micro-Raman spectroscopy[J]. Journal of Raman Spectroscopy, 2017, 48(12): 1871-1880. doi: 10.1002/jrs.5261 [26] SUN H, LIANG H, ZHOU G, et al. Supported cobalt catalysts by one-pot aqueous combustion synthesis for catalytic phenol degradation[J]. Journal of Colloid and Interface Science, 2013, 394: 394-400. doi: 10.1016/j.jcis.2012.11.017 [27] YANG Q, CHOI H, DIONYSIOU D D. Nanocrystalline cobalt oxide immobilized on titanium dioxide nanoparticles for the heterogeneous activation of peroxymonosulfate[J]. Applied Catalysis B:Environmental, 2007, 74(1/2): 170-178. doi: 10.1016/j.apcatb.2007.02.001 [28] WANG J, WANG S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. doi: 10.1016/j.cej.2017.11.059 [29] HUANG Y H, HUANG Y F, HUANG C I, et al. Efficient decolorization of azo dye Reactive Black B involving aromatic fragment degradation in buffered Co2+/PMS oxidative processes with a ppb level dosage of Co2+-catalyst[J]. Journal of Hazardous Materials, 2009, 170(2-3): 1110-1118. doi: 10.1016/j.jhazmat.2009.05.091 [30] QI C, LIU X, LIN C, et al. Degradation of sulfamethoxazole by microwave-activated persulfate: Kinetics, mechanism and acute toxicity[J]. Chemical Engineering Journal, 2014, 249: 6-14. doi: 10.1016/j.cej.2014.03.086 [31] ZHANG Q, CHEN J, DAI C, et al. Degradation of carbamazepine and toxicity evaluation using the UV/persulfate process in aqueous solution[J]. Journal of Chemical Technology & Biotechnology, 2015, 90(4): 701-708. [32] AO X, LIU W. Degradation of sulfamethoxazole by medium pressure UV and oxidants: Peroxymonosulfate, persulfate, and hydrogen peroxide[J]. Chemical Engineering Journal, 2017, 313: 629-637. doi: 10.1016/j.cej.2016.12.089 [33] BUXTON G V, BYDDER M, SALMON G A, et al. The reactivity of chlorine atoms in aqueous solution. Part III. The reactions of Cl• with solutes[J]. Physical Chemistry Chemical Physics, 2000, 2(2): 237-245. doi: 10.1039/a907133d [34] YAN J, LI J, PENG J, et al. Efficient degradation of sulfamethoxazole by the CuO@Al2O3 (EPC) coupled PMS system: Optimization, degradation pathways and toxicity evaluation[J]. Chemical Engineering Journal, 2019, 359: 1097-1110. doi: 10.1016/j.cej.2018.11.074 [35] WANG S, LIU H, WANG J. Nitrogen, sulfur and oxygen co-doped carbon-armored Co/Co9S8 rods (Co/Co9S8@N-S-O-C) as efficient activator of peroxymonosulfate for sulfamethoxazole degradation[J]. Journal of Hazardous Materials, 2020, 387: 121669. doi: 10.1016/j.jhazmat.2019.121669 [36] LAI L, YAN J, LI J, et al. Co/Al2O3-EPM as peroxymonosulfate activator for sulfamethoxazole removal: Performance, biotoxicity, degradation pathways and mechanism[J]. Chemical Engineering Journal, 2018, 343: 676-688. doi: 10.1016/j.cej.2018.01.035 [37] AO X, LIU W, SUN W, et al. Mechanisms and toxicity evaluation of the degradation of sulfamethoxazole by MPUV/PMS process[J]. Chemosphere, 2018, 212: 365-375. doi: 10.1016/j.chemosphere.2018.08.031 [38] YANG S, WANG P, YANG X, et al. Degradation efficiencies of azo dye Acid Orange 7 by the interaction of heat, UV and anions with common oxidants: Persulfate, peroxymonosulfate and hydrogen peroxide[J]. Journal of Hazardous Materials, 2010, 179(1-3): 552-558. doi: 10.1016/j.jhazmat.2010.03.039 [39] JI Y, XIE W, FAN Y, et al. Degradation of trimethoprim by thermo-activated persulfate oxidation: Reaction kinetics and transformation mechanisms[J]. Chemical Engineering Journal, 2016, 286: 16-24. doi: 10.1016/j.cej.2015.10.050 [40] WANG C, KIM J, KIM M, et al. Nanoarchitectured metal-organic framework-derived hollow carbon nanofiber filters for advanced oxidation processes[J]. Journal of Materials Chemistry A, 2019, 7(22): 13743-13750. doi: 10.1039/C9TA03128F [41] LIN K Y A, LIN T Y, LU Y C, et al. Electrospun nanofiber of cobalt titanate perovskite as an enhanced heterogeneous catalyst for activating peroxymonosulfate in water[J]. Chemical Engineering Science, 2017, 168: 372-379. doi: 10.1016/j.ces.2017.05.013 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4620
- HTML全文浏览数: 4620
- PDF下载数: 140
- 施引文献: 0
