

 下载:
下载:

-
在水处理技术中,混凝是一种常见的工艺,其可以与许多其他处理方法(膜滤、氧化等)相结合,从而达到净水目的[1]。近年来,不同的混凝变量(混凝剂的类型和浓度)以及絮体的特性(再生强度和再生能力)受到了相当大的关注[2-4]。铝盐和铁盐均为常见的混凝剂,与铝盐混凝剂相比,铁盐混凝剂可以在更宽的pH和温度范围内使用,而且铁盐更容易生成较大的絮体,更利于沉降分离[5-6]。在混凝过程中,Fe(Ⅱ)/Fe(Ⅲ)盐会快速水解成铁氢氧化物纳米颗粒,而后聚集形成絮体沉淀,而且随着纳米颗粒的聚集和结晶,其结构也会发生变化[7-8]。大量研究表明,铁(氢)氧化物的结晶和转化,会显著影响污染物(磷、砷等)的迁移[9-10]。比如,老化结晶过程会导致絮体的比表面积变小,进而导致其去除磷酸盐的效率变低。结晶和转化过程一般可以用2种不同的生长模式来描述:一种是奥斯瓦尔德熟化,其描述了一种非均匀结构随时间所发生的变化,溶质中的小型晶体或溶胶颗粒溶解并再次沉积到大型的晶体或溶胶颗粒上;另一种是定向附着机制,体系中的微晶通过旋转合适的角度附着到沿相同晶向生长的较大晶体的晶面上,最终聚集体可以看作是原始晶体颗粒以不可逆和高度统一取向的方式构建而成的大单晶[11-14]。
Fe盐水解沉淀及其后续结晶转化过程会受到有机质、pH、无机盐和光照等环境条件的影响[15] 。其中,有机质对Fe沉淀的转化受到了广泛的关注,具有不同分子质量、不同官能团和含量的有机质对沉淀的形成和转化有不同的影响。许多研究[16-17]表明,在混凝过程中有机物所含的羧基官能团是其重要的反应位点。
然而,小分子有机物如何影响混凝过程,目前还没有明确或充分的解释。虽然小分子有机物通常不是混凝去除的目标污染物,但其对混凝性能的影响(颗粒聚集时间、絮体大小和结晶度等)不容忽视。因此,本研究选用有2个羧基和1个氨基的天冬氨酸,以亚铁盐(Fe(Ⅱ))作为混凝剂,探讨了不同浓度的天冬氨酸对絮体形成过程和对磷的去除效果的影响。
-
七水合硫酸亚铁(Ⅱ) (FeSO4·7H2O,CAS:7782-63-0)、碳酸氢钠(NaHCO3,CAS:144-55-8)、四水钼酸铵(4MoO3·3H2MoO4·4H2O·6H3N,CAS:12054-85-2)、抗坏血酸(C6H8O6,CAS:50-81-7)、浓硫酸(H2SO4,CAS:7664-93-9)和磷酸二氢钾(KH2PO4,CAS: 7758-11-4)均购自国药化学试剂有限公司(中国上海),天冬氨酸(Asp, Aspartic acid, CAS: 6899-03-2)购自百灵威。所有试剂均为分析级。
-
混凝实验中,在含0.42 g碳酸氢钠的纯水中分别加入不同浓度的天冬氨酸(Asp),超声至完全溶解并定容至1 L,最终制得浓度分别为0、0.4、1、1.5、2.5、5和10 mmol·L−1梯度的天冬氨酸溶液,然后使用稀盐酸(HCL, 0.2 mol·L−1)或氢氧化钠溶液(NaOH, 0.2 mol·L−1)将所得溶液调至pH为7,溶液中的溶解氧使用便携式溶解氧仪(F4, METTLERTOLEDO, Switzerland, with LE621 IP67 dissolved oxygen sensor)测定。使用硫酸亚铁(0.1 mmol·L−1)作为混凝剂,通过混凝装置(MY3000-2N,武汉梅宇,中国)控制混凝体系中絮体的生长和破碎。在此过程当中用光度分散分析仪Photometric Dispersion Analyzer (PDA2000, Rank Brothers Ltd,UK)对絮体的生长情况进行实时监测,该仪器通过连续循环检测水中颗粒的变化和波动;每秒收集均方根(rms)和平均透射光强(dc)的比值。rms与dc的比值称为絮凝指数(FI), FI可以反映絮凝体在絮凝实验过程中的实时生长情况。
投加0.1 mmol·L−1的FeSO4絮凝剂后,以200 r·min−1的速度快速搅拌1 min使溶液混合均匀,而后以50 r·min−1 的速度缓慢搅拌观察絮体生长情况;当絮体生长至平稳值持续500 s后以200 r·min−1的速度对絮体进行1 min的剪切破碎,然后恢复转速至50 r·min−1使絮体再次生长,直至所得絮体的大小不再发生明显变化终止实验。光度色散分析仪连接电脑的bubble4软件上实时记录检测所得数据,PDA的读数大小不再发生较大波动时,认为絮体生长完成。所有的混凝实验均在室温(25±1) ℃下进行。
磷的去除实验中,称取0.252 g二水合磷酸二氢钠 (NaH2PO4·2H2O)于100 mL容量瓶中加纯水定容至刻度线作为磷酸储备液(0.5 g·L−1)。在混凝实验中絮体生长稳定至峰值时,取1 mL磷酸储备液加入到1 L的混凝体系当中,此时磷酸盐的初始质量浓度为0.5 mg·L−1,以50 r·min−1的速度搅拌10 min后用0.45 μm的微孔滤膜(聚偏二氟乙烯(PVDF)、直径50 mm、津腾)进行固液分离,液相用来测定反应后磷酸盐的浓度,絮体用来进行表面官能团、成分和结晶度分析。
-
在混凝实验中,当絮凝指数(FI, flocculation index)达到稳定的峰值时,通过使用0.45 μm微滤膜进行抽滤将所得絮体沉在膜表面,然后将其冷冻后进行真空冷冻干燥,经过约48 h后将完全冷干的样品取出。对冷干之后的絮体样品进行如下表征:采用扫描电镜(Hitachi SU8020, Japan)观察絮体的生长和表面形貌变化;采用透射电镜(Tecnai G2 F30 S-TWIN)观察絮体的微观形貌,鉴定其物相和结晶度;采用X射线衍射仪 (XRD, X’Pert PRO MPD, PAN alytical)在40 kV、40 mA的条件下对其进行测定分析,以获得材料的结晶程度、成分及其所占比例和晶体结构信息,(衍射角为5~90°,扫描速度为6o·min−1)。使用x射线光电子能谱仪(XPS, PHI Quantera II, ULVAC, Japan)在150 W Al-K α辐射条件下测定样品的化学状态信息,不同元素的结合能由C1s校准 (284.8 eV)。傅里叶红外变换光谱(FTIR)被用于分析冷冻干燥后絮体的表面官能团性质及相对含量(扫描范围为400~4 000 cm−1)。
在磷的去除实验中,磷的测定采用钼酸铵分光光度法(GB 11893-89)测定,通过钼酸铵与正磷酸盐反应生成黄色磷酸杂多酸,而后被抗坏血酸还原成磷钼蓝,于700 nm处用分光光度计测其吸光度,带入标准曲线计算出溶液中的磷酸盐含量。
-
PDA结果如图1所示。可见,在DO为5 mg·L−1、pH为7的混凝体系中添加天冬氨酸后,絮体的颗粒尺寸显著增大,随着体系中有机物浓度的增加,絮体生长的滞后时间延长,由于排列在絮体外侧的铁离子率先水解,天冬氨酸与—OH竞争占据铁离子表面的活性位点,从而改变铁离子在水中的形态。加入天冬氨酸等同于在体系中加入了大量带负电荷的物质,提高了絮凝指数,由于天冬氨酸的桥接作用,絮体的尺寸相较于不添加天冬氨酸时均有明显的增加,当天冬氨酸的浓度为1.5 mmol·L−1时,絮体的尺寸在2 500 s左右达到峰值,絮体的FI指数由1.5%左右上升到3%,说明此时絮凝后絮体会更大。破碎后絮体可以再生长到更接近于破碎前的大小。这可能是由于氢氧根沉淀物内的化学键仅有少量断裂、未引起絮体表面性质改变[18],更有利于絮体的沉淀。随着天冬氨酸的浓度继续增加,可能由于天冬氨酸的静电斥力和空间位阻作用占主导地位,导致絮体的FI指数开始减少直至无明显的絮体生长。在混凝过程中,当搅拌速率从50 r·min−1增加到200 r·min−1时,絮体破碎后,纳米颗粒表面活性减小(或失去),致使絮体无法生长至破碎前的尺寸[18-19]。
-
在DO为5 mg·L−1、pH为7的条件下,分别添加0、0.4、1、1.5、2.5、10 mmol·L−1天冬氨酸,当絮体生长且稳定到最大FI值时,加入0.5 g·L−1磷酸盐储备液1 mL于混凝杯中,此时体系中的磷酸盐初始值为0.5 mg·L−1,磷酸盐去除效果如图2所示。结果表明,在不添加天冬氨酸时,去除磷酸盐后其质量浓度保持在0.2 mg·L−1;添加天冬氨酸混凝后磷酸盐残余质量浓度降低至0.02 mg·L−1,有效提高了磷酸盐的去除效果。经过计算在不添加天冬氨酸时生成絮体对磷酸盐的去除率为63.16%。在添加天冬氨酸浓度为0.4、1、1.5、2.5 mmol·L−1时,生成的絮体对于磷酸盐的去除率分别为91.96%、97.26%、93.9%、99.6%,而当天冬氨酸的浓度为10 mmol·L−1时,磷酸盐的去除率仅有52.91%。这是由于过多的天冬氨酸不仅在絮体的形成过程中起到了桥接作用,而且在后续的磷酸盐去除阶段也占据了絮体表面的活性位点与其形成了竞争吸附的关系,从而导致了对于磷酸盐的去除效果不理想。
-
在DO为5、pH为7时絮体表面形貌的SEM表征结果如图3(a)所示。由图3(a) 可见,无添加天冬氨酸体系内,絮体呈现出50 nm左右的小球和100 nm大小的片状混合体;如图3(b)所示,和没有添加天冬氨酸相比,添加1.5 mmol·L−1天冬氨酸后絮体尺寸变大(约200 nm),这与PDA的结果一致。如图3(c)所示,当天冬氨酸浓度增加到2.5 mmol·L−1时,絮体的纳米颗粒相互连接聚拢,呈现出更加清晰的褶皱状结构。
-
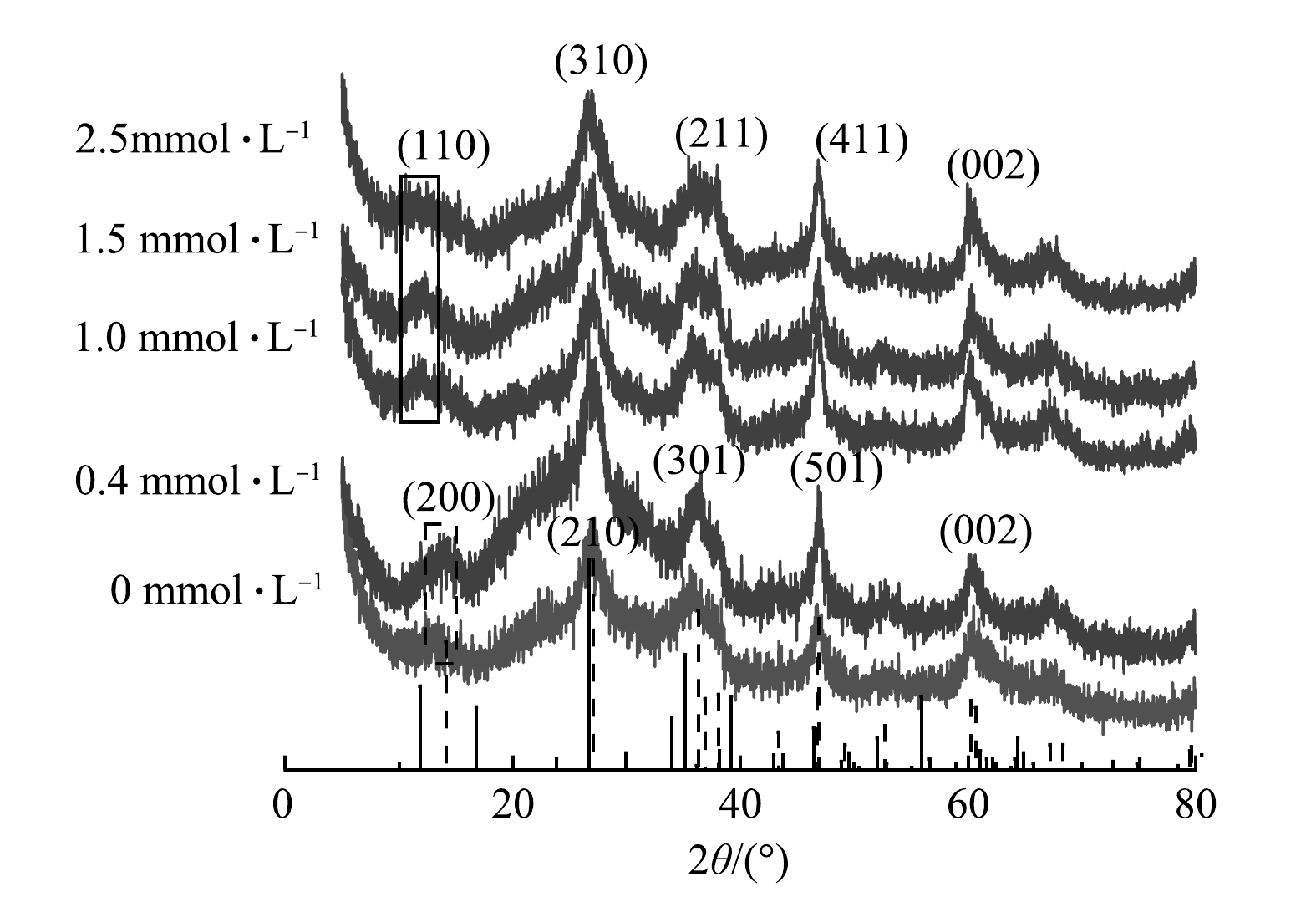
如图4所示,在不添加天冬氨酸和添加0.4 mmol·L−1天冬氨酸时,所对应的XRD图中2θ为14.113°、27.047°、39.296°、46.778°、52.714°、67.218°处,鉴定为纤铁矿(JCPDS card 44-1415),晶格常数a为12.52 Å、b为3.873 Å、c为3.071 Å。添加0.4 mmol·L−1 天冬氨酸与不添加天冬氨酸相比,结晶度明显增强。这可能是由于天冬氨酸诱导了其矿物的转化,使其具有更加良好的晶型。此外磷酸盐去除率显著提升,说明了少量的天冬氨酸的存在间接促进了磷酸盐的去除。这可能是由于天冬氨酸中的羧基促进絮体向纤铁矿(γ-FeOOH)的转化,为磷酸盐提供了更多去除位点[20]。当体系中天冬氨酸的浓度为1 mmol·L−1及以上时,絮体所对应的衍射峰位于2θ为11.842°、26.725°、35.161°、46.433°、61.097°处,这表明絮体的主要成分为四方纤铁矿(JCPDS card 34-1266),晶格常数a为10.535 Å、b为10.535 Å、c为3.03 Å。值得注意的是,当体系中天冬氨酸的浓度为小于0.4 mmol·L−1时,随着天冬氨酸浓度增加絮体结晶性增加,然而当天冬氨酸浓度增加到1 mmol·L−1,絮体的结晶度降低。这可能是由于在低C/Fe比时,有机物在与铁离子络合在一起,对亚铁离子转化为三价的羟基氧化铁有促进作用[21-22];然而在体系中有较高浓度的有机物时,纳米颗粒的表面活性位点更多的被封锁,限制了纳米颗粒之间的吸附,同时有机物的静电排斥和空间位阻的影响阻止纳米颗粒的聚集,因此,限制了羟基氧化铁的生长[23],导致絮体结晶性降低。
为了提供γ-FeOOH和β-FeOOH存在的直接证据,采用HR-TEM对pH为7、DO为 5.0 mg·L−1、含1.5 mmol·L−1 天冬氨酸混凝后的絮体进行了细致的研究。如图5(a)所示,絮体在纳米尺度下的形貌与SEM(图3(b))下的形貌皆呈现出较大的板片状,密集的堆叠在一起形成大的聚集体,图5(a)中区域A如图5(b)所示,分别对应β-FeOOH (004)和γ-FeOOH (-101)的晶格面,晶面间距为2.606 Å和3.048 Å;图5(a)中区域B如图5(c)所示,对应于β-FeOOH (004) 晶面,晶面间距为2.62 Å,与图5(d)中晶胞结构图分别对应。
-
采用XPS和FTIR对pH为7、DO为5.0 mg·L−1、含1.5 mmol·L−1 天冬氨酸混凝后的絮体进行了表面官能团分析。如图6(a)所示,Fe2p的XPS光谱中含有2个主峰,分别为Fe2p3/2(711.4 eV),Fe2p1/2(725.4 eV),连同2个重要的卫星峰,其分别在720.1 eV和733.1 eV,此外,未发现有其他杂质峰的存在。结合O光谱图6(b),进一步证实了Fe—O—C键的存在。O1s可以分为Fe—O(531.2 eV)、Fe—O—C(532.4 eV)、Fe—OH(533.1 eV) 3个峰,说明了絮体的表面的确形成了羟基氧化铁。如图5(c)所示,C1s峰显示有3个峰,分别为C—C(284.1 eV)、C—O(286.6 eV)、C=O(288.7 eV),证实了天冬氨酸存在于絮体的表面,与絮体形成了络合物。进一步对絮体进行了FTIR分析,结果如图6(d)所示。3 177 cm−1处归属于羟基峰[24-25],在1 642 cm−1处的峰是由于晶体结构中的H2O的震动引起的,1 020 (面内弯曲振动)、746 (面外弯曲振动)、476 cm−1对应于Fe—O—H不同的弯曲振动[26]。结合XRD的表征结果,可进一步证明γ-FeOOH和β-FeOOH的存在。
-
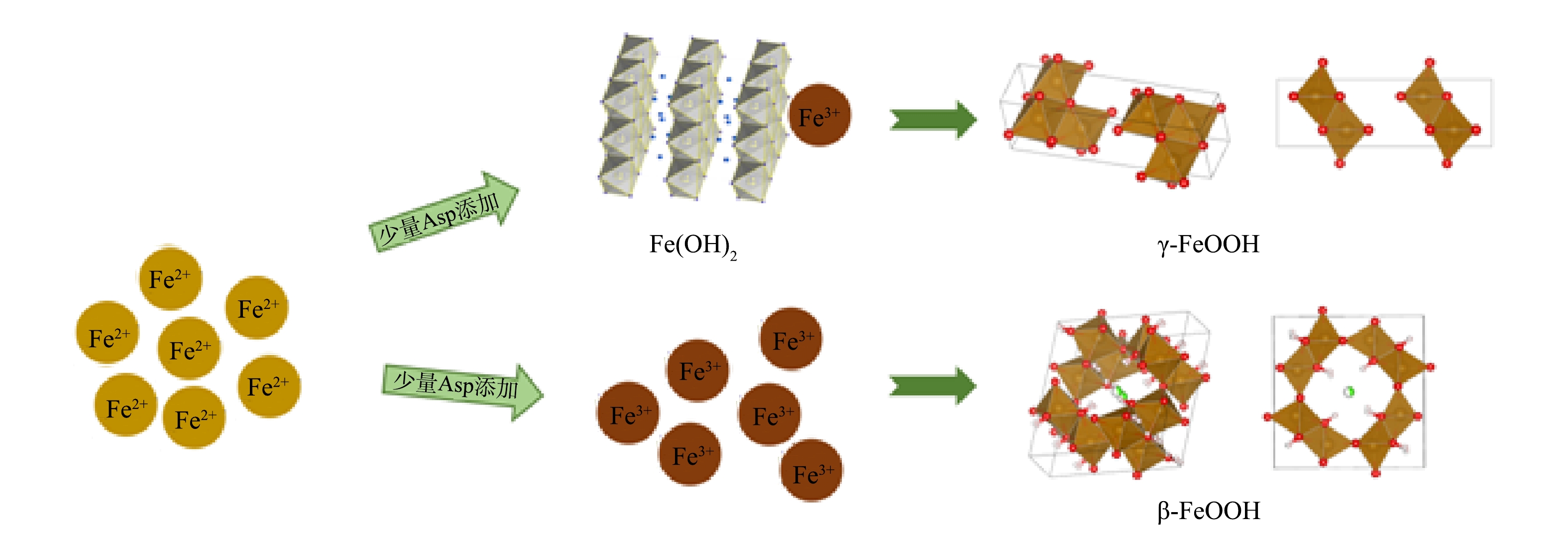
综上所述,Fe(Ⅱ)混凝中产生了形貌相似的片状絮凝体,但其主要成分存在差异。当体系中存在少量天冬氨酸时,由于Fe(Ⅱ)能在水溶液中快速水解,体系中部分Fe(Ⅱ)会被包裹,Fe(OH)2纳米颗粒由于铁化合物的部分饱和快速形成,随后作为晶核外延生长,Fe(OH)2纳米离子表面被氧化转化成三价铁,当溶液颜色完全变黄时,最终形成γ-FeOOH或者β-FeOOH(图7)。此外,少量的天冬氨酸添加明显增加了絮体γ-FeOOH的结晶性(图4)。然而当天冬氨酸的浓度增加到0.4 mmol·L−1以上时,絮体的主要成分不再是γ- FeOOH而是β-FeOOH。这可能是由于增多的有机物会将部分亚铁离子完全包裹,导致剩余的亚铁离子被快速氧化为三价铁,在天冬氨酸的作用下转化成了β-FeOOH。
-
1)在pH为7,DO为5 mg·L−1的条件下,添加天冬氨酸可使絮体的生长显著延后,不同浓度天冬氨酸的添加使絮体的形成过程存在差异从而影响絮体的主要成分。添加0.4 mmol·L−1以下天冬氨酸时,Fe(OH)2纳米颗粒由于铁化合物的部分饱和快速形成,随后作为晶体的核心外延生长,此时絮体的主要成分是γ- FeOOH;在添加大于0.4 mmol·L−1时,未被天冬氨酸完全包裹的Fe(Ⅱ)被迅速氧化,同时,在天冬氨酸的作用下絮体的主要成分是β-FeOOH。
2)添加1.5 mmol·L−1天冬氨酸时的磷酸盐去除率93.9%,此时絮体的FI指数最高,絮体更加容易沉降分离。
3)相较于不添加天冬氨酸生成的絮体,经过天冬氨酸诱导后形成的絮体对磷酸盐去除能力约为前者的1.57倍,磷酸盐的质量浓度降至0.02 mg·L−1,有效降低了富营养化的累积。
天冬氨酸对亚铁混凝剂絮体结构及磷酸盐去除效果的影响
Effect of aspartic acid on the floc composition of Fe(II) coagulant and its deep phosphorus removal
-
摘要: 铁混凝剂广泛应用于水处理领域,形成的絮体大小、形态和结晶度直接决定其混凝效果。然而小分子有机物对混凝的影响机制尚不清楚。本研究以FeSO4作为混凝剂,通过添加特定的小分子有机物(分子质量<1 000 Da)天冬氨酸,研究了天冬氨酸对絮体生长过程及其产物的影响。结果表明,天冬氨酸通过影响金属的水解和纳米颗粒的性质进而影响其混凝性能。在pH=7时,天冬氨酸的存在延缓了絮体的初始生长时间,但增大了絮凝体的最大粒径(从0.05 μm大小的碎片和小球状参杂的絮体变化至0.1 μm的褶皱状絮体)和磷酸盐的去除率。当添加0.4 mmol·L−1的天冬氨酸时,纤铁矿(γ-FeOOH)为絮体的主要成分,这可能少量的天冬氨酸存在时,体系中部分亚铁离子会被包裹, Fe(OH)2纳米颗粒由于铁化合物的饱和快速形成,随后作为晶核生长; Fe(OH)2纳米颗粒表面通过被氧化转化成三价铁,最终形成γ-FeOOH;当添加天冬氨酸大于0.4 mmol·L−1时,絮体的主要成分为四方纤铁矿(β-FeOOH),此时部分亚铁离子被完全包裹,剩余的亚铁离子被氧化成三价铁后形成β-FeOOH。经过天冬氨酸诱导后形成的絮体对磷酸盐的去除率均增加了1.57倍左右,处理后的磷酸盐浓度降低到了0.02 mg·L−1,且在添加1.5 mmol·L−1 天冬氨酸时絮体的尺寸最大,此时的絮体更加容易沉降分离。Abstract: Iron flocculants are widely used in water treatment applications where the size, morphology, and crystallinity of the flocs directly determine the adsorption activity and settling performance. However, the effect mechanism of small molecular organics on coagulation is still unclear. In this study, FeSO4 was used as coagulant, its effects of adding specific small molecule organic matter (molecular weight <1000 Da) of aspartic acid (containing two carboxyl groups and one amino group) on flocs growth process and products were investigated. The results show that aspartic acid affected metal hydrolysis, nanoparticles properties and the coagulation performance. At pH=7, the presence of aspartic acid delayed the initial growth time of flocs, but increased the maximum particle size of flocs, changing from 0.05 μm sized flocs with fragments and small spherical participations to 0.1 μm folded flocs, and the phosphate removal rate. When 0.4 mmol·L−1 aspartic acid was added, lepidocrocite (γ-FeOOH) was the main component of the floc. Due to the rapid hydrolysis of Fe(Ⅱ) ions in aqueous solution, when there was a small amount of aspartic acid in the system, part of the Fe(Ⅱ) ions in the system would be wrapped, and Fe(OH)2 nanoparticles would form rapidly due to the saturation of iron compounds. Afterwards, Fe(OH)2 nanoparticles continued to grow as the crystal, its surface was oxidized into Fe(Ⅲ), finally γ-FeOOH formation occurred. When the dosage of aspartic acid was higher than 0.4 mmol·L−1, the main component of flocs was akaganeite (β-FeOOH). At this time, part of Fe(Ⅱ) ions were completely wrapped, and the remaining Fe(Ⅱ) ions were oxidized into Fe(Ⅲ) irons and hydrolyzed to form β-FeOOH. The phosphorus removal rate by the flocs induced by aspartic acid increased by about 1.57 times, and the phosphate concentration decreased to 0.02 mg·L−1 after treatment. Moreover, the FI index of flocs was the highest when 1.5 mmol·L−1 aspartic acid was added, and the flocs were easier to settle and separate.
-
Key words:
- aspartic acid /
- floc /
- lepidocrocite and akageneite /
- phosphorus removal
-
土壤和地下水中铬污染是我国当前亟待解决的环境问题。铬污染主要来源于铬盐生产、电镀、制革等工业废水及固体废弃物的排放[1]。历史上,我国曾有大量露天堆积的铬渣,虽然经整治与处理后大部分铬渣已得到安全处置[2],但铬渣堆场附近仍存在大面积受Cr(Ⅵ)污染土壤[3]。这些受Cr(Ⅵ)污染的土壤,由于淋溶作用,极易溶出Cr(Ⅵ),导致场地周围地下水在相当长的时间内受到Cr(Ⅵ)潜在污染的威胁[4-6]。因此,开展土壤-地下水Cr(Ⅵ)污染修复具有重要的实际意义。
自然条件下,铬在土壤-地下水中主要以Cr(Ⅲ)和Cr(Ⅵ) 2种氧化状态存在[7]。其中,Cr(Ⅲ)在pH=6~12时,由于能够形成稳定的氢氧化物沉淀析出[8],导致其迁移性大大降低;同时Cr(Ⅲ)毒性较低,故仅在很高浓度条件下才表现出生物毒性[9]。与此相反,Cr(Ⅵ)具有致癌性,因易溶于水,故在地下水中具有很强的迁移性[10, 11]。因此,对受Cr(Ⅵ)污染的地下水常采用原位还原固定化技术,通过向地下含水层中注入还原剂将Cr(Ⅵ)还原固定为Cr(Ⅲ)降低其迁移性和毒性,从而实现修复的目的。
常用的Cr(Ⅵ)原位修复技术主要包括可渗透反应墙(PRB)、生物修复、电动修复和原位氧化还原控制墙技术(in situ redox manipulation, ISRM)等。传统的PRB技术通过在污染源下游地下含水层中开挖沟槽、回填活性反应材料以实现修复目的。但实际应用中该技术一般用于受污染的浅层地下水,不适于深部地下水的修复。ISRM技术是近年来提出的一类新型的原位还原技术。该技术通过注入井向地下污染区域注入还原剂以建立还原性环境,从而实现污染物的还原修复[12-15]。与PRB技术相比,ISRM对较深含水层的污染修复具有明显的优势。
地下水修复中常用的还原剂包括硫类化合物、铁基及有机类还原剂[16]。其中硫基还原剂因具有优异的反应活性,在地下水Cr(Ⅵ)还原固定化修复中受到广泛的关注。采用ISRM技术修复受Cr(Ⅵ)污染地下水中研究最多的2种硫化物为连二亚硫酸钠和多硫化物。虽然连二亚硫酸钠具有很强的还原能力,但由于其在中性水溶液中不稳定,在实际修复中需要外加碱性物质(如碳酸钾/碳酸氢钾缓冲)[14],因此,在实际应用中受到一定限制。有研究表明,多硫化物(Sx2-)性能稳定、还原能力强,可以在多孔介质中形成相对持久的还原性区域,是EPA推荐的修复试剂,近年来,在地下水修复中引起了广泛的关注[12,17]。连二亚硫酸钠与多硫化物的修复机理类似。以ISRM中采用多硫化物还原固定化Cr(Ⅵ)为例,通过注入井向含水层注入的多硫化物首先还原孔隙水中的Cr(Ⅵ)使其以三价铬的形式固定下来;而剩余的多硫化物能进一步与含水层中含铁矿物中的Fe(Ⅲ)反应生成Fe(Ⅱ)。SHI等[18]的研究表明,多硫化物可以与纤铁矿反应生成含二价铁的次生还原性产物;所产生的Fe(Ⅱ)以固相和强吸附态形态为主,而溶解态的相对较少。这也解释了多硫化物具有在受污染含水层中建立持久还原性区域能力的原因,使其在地下水还原修复中得到广泛研究和应用[13,19]。
现场实验结果也表明,多硫化物对受铬污染土壤地下水的修复具有优异的效果[1,20-23]。一项有关采用多硫化钙对英国格拉斯哥某受铬渣污染地下水进行的中试研究结果表明:在所选用的3种修复方法中,以直接向含水层注入多硫化钙的方案效果最佳,可以将Cr(Ⅵ)浓度从800 μg·L−1降至30 μg·L−1。MESSER等[17]对美国亚利桑那某电镀污染现场的中试结果表明,经多硫化物处理,可使包气带土壤和孔隙水中Cr(Ⅵ)分别从处理前的2 190 mg·kg−1和3 600 mg·L−1降至10%以下,而含水层中的Cr(Ⅵ)迅速由240 mg·L−1降至1 mg·L−1。进一步的检测表明,多硫化物在含水层中所形成的还原性区域能维持数月,对地下水中Cr(Ⅵ)污染具有长期修复的潜力。
由于褐土和红壤是我国2种重要的土壤类型,其分别在河南省和云南省有广泛的分布,而且在两省均曾有铬渣的堆放,因此,本研究选择褐土和红壤为研究对象,在室内通过土壤柱实验模拟研究了采用ISRM技术还原固定化修复受Cr(Ⅵ)污染的土壤-地下水的过程,考察了多硫化物对土壤样品的硫化处理及硫化后对Cr(Ⅵ)的还原能力,以及还原Cr(Ⅵ)柱实验过程中多硫化物的电子利用效率等。在柱实验过程中,同时对出水pH、氧化还原电位进行实时监测,以考察柱体内是否有效地建立了还原区域。研究结果可对以多硫化物为还原剂采用ISRM技术现场修复Cr(Ⅵ)污染的土壤-地下水具有一定的参考价值。
1. 材料与方法
1.1 多硫化物的制备
将0.1 mol·L−1硫化钠溶液与等摩尔数的单质硫加入500 mL血清瓶中,转移至恒温振荡箱中,在65 ℃和100 r·min−1条件下振荡16 h[24-25]。多硫化物和硫化钠浓度采用碘量法[26]测定。
1.2 土壤样品的采集和预处理
实验所用的红壤和褐土样品分别采自云南和河南。样品采集使用棋盘法,首先在采样现场选取7~10个采样点,在地表下10~30 cm的土层,每个采样点取2~3 kg样品。采集的土壤样品在实验室通风阴凉处风干并剔除土壤中杂质,用木锤敲打风干的大块样品,压碎并混匀后研磨过2 mm尼龙筛,筛下物样品用于土柱实验。
1.3 室内土壤柱实验模拟研究
图1(a)为所搭建的实验室土壤柱实验装置。柱实验过程中采用自下而上的入水方式。实验中所有溶液均采用经氮气吹脱的Milli-Q超纯水(18.2 MΩ)配制,以保证整个实验过程在厌氧条件下进行。
 图 1 土壤柱实验装置与土柱结构示意图Figure 1. Schematic diagrams of the column experiment setup and the structure of the soil-packed column
图 1 土壤柱实验装置与土柱结构示意图Figure 1. Schematic diagrams of the column experiment setup and the structure of the soil-packed column土柱装填采用干堆法[27-28]。在装填过程中,每次加入等量的样品,用橡胶锤轻轻击打柱体并均匀压实,以免颗粒间产生明显缝隙,从而使实验中所使用柱子性质基本保持一致。在实验运行过程中,若土壤的黏性较大可能会导致柱内土壤无法均匀润湿、柱压过高,从而造成土柱中的优先流或柱子堵塞等现象。此时可在土壤中掺杂质量比1∶1的石英砂(0.1~0.4 mm)[29],以避免上述现象的出现。图1(b)为按上述方法所装填好的土柱,土柱内径和装填高度分别为2.5 cm和20 cm。装填好的红壤柱和褐土柱的孔隙率分别为0.50和0.49,所装填土壤质量分别为130.3 g和136.3 g。
土壤柱实验包括土柱预处理、土柱硫化以及硫化土柱对Cr(Ⅵ) 还原固定化3个过程。在土柱预处理中,向柱体通入模拟地下水以充分润湿土壤并使孔隙饱和。实验中所用模拟地下水组成[30]为10 mmol·L−1 Na+、0.8 mmol·L−1 Ca2+、6.6 mmol·L−1 Cl−、3 mmol·L−1
HCO−3 、1 mmol·L−1SO2−4 。在土壤硫化过程中,向上述经水饱和的土柱以1.15 mL·min−1流速通入初始浓度为50 mmol·L−1多硫化物,并在设定时间内对出水口处溶液的pH、氧化还原电位和多硫化物浓度进行测定。当多硫化物浓度接近50 mmol·L−1时,停止通入含多硫化物的地下水。在接下来的实验中,向硫化后土柱以同样流速通入含有10 mg·L−1 Cr(Ⅵ)的模拟地下水,以观测硫化土柱对Cr(Ⅵ)的持续还原能力。在实验中,在既定时间取出水口处样品记录溶液的pH和氧化还原电位,同时分析多硫化物和Cr(Ⅵ)的浓度。基于我国地下水分类标准(GB/T 14848-2017),针对Cr(Ⅵ)这一指标,Ⅳ类和Ⅴ类的界限为0.1 mg·L−1,本实验将出水中Cr (Ⅵ)的浓度达0.1 mg·L−1时定义为穿透点,达到穿透点后,继续通入Cr(Ⅵ) 模拟地下水,当出水浓度接近10 mg·L−1时,停止柱实验。
土壤柱实验过程中多硫化物浓度的测定采用亚甲基蓝分光光度法[31],Cr(Ⅵ)浓度的测定采用《水质 六价铬的测定 二苯碳酰二肼分光光度法》(GB/T 7467-1987),pH的测定使用pH电极(InLab Expert Go-ISM, METTLER TOLEDO),氧化还原电位的测定使用氧化还原电极(ORION 9678BNWP, Thermo)。
2. 结果与讨论
2.1 土柱的硫化处理
图2(a)和图2(b)分别为红壤和褐土柱子在多硫化物硫化处理过程中,出水溶液的pH、氧化还原电位以及多硫化物浓度随时间的变化规律。实验中所观察到的红壤和褐土柱硫化现象类似。
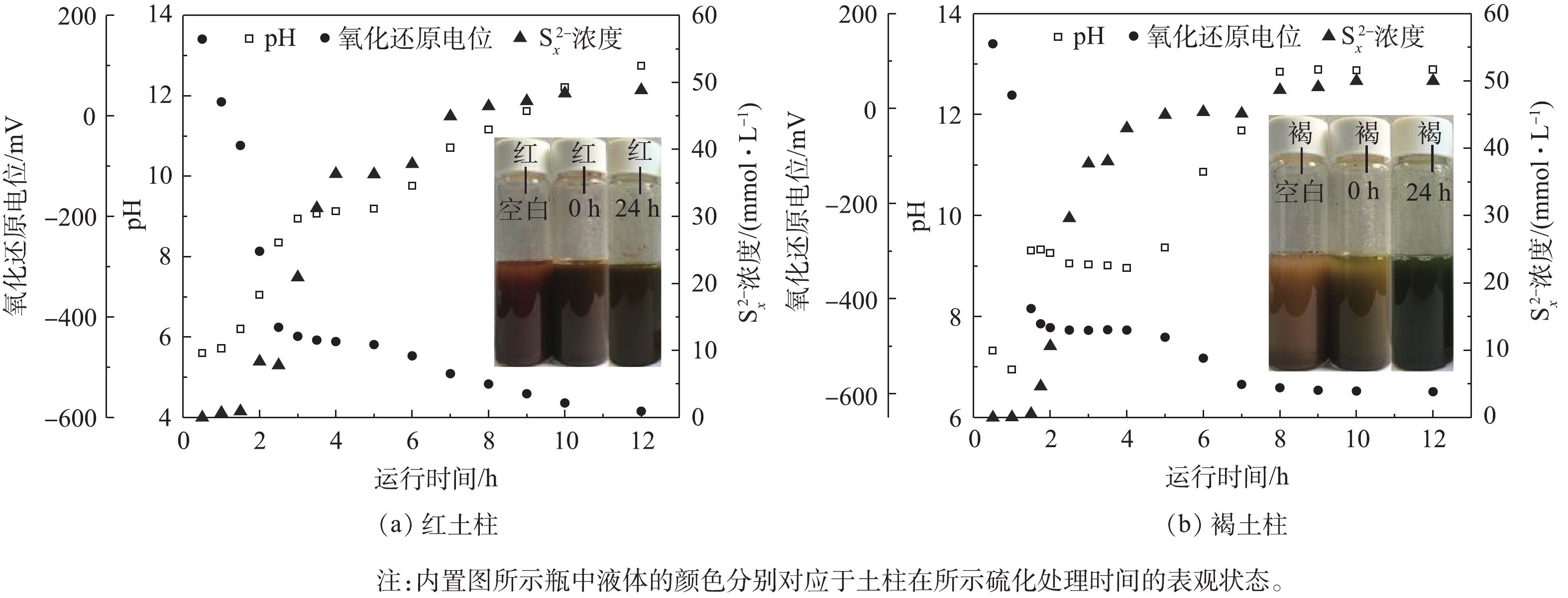 图 2 出水溶液的pH、氧化还原电位及
图 2 出水溶液的pH、氧化还原电位及S2−x 浓度随时间的变化Figure 2. Changes of pH, oxidation-reduction potential andS2−x concentrations in outlet solutions with time在2种土柱硫化过程中均观察到出水和柱体颜色随时间发生了变化。对于红壤实验,当向红壤柱中通入约1~2个PV多硫化物后,出水溶液开始呈现乳白色,这可能是由于单质硫的析出所造成的。之后出水颜色逐渐加深,并最终呈深墨绿色,而柱内的土壤由初始的红色自下而上渐渐呈现出深墨绿色。在褐土柱硫化过程中,颜色随时间的变化与红壤柱变化规律类似,出水溶液的颜色逐渐加深并最终变为草绿色;土柱内土壤从初始的棕褐色由下至上最终全部变为绿色。2种土柱及其出水颜色随时间发生变化可能是由于土壤中含铁物质与多硫化物反应,生成了亚铁以及硫铁化合物所造成[12,18,32-33]。
在红壤柱和褐土柱的硫化过程中也观察到了出水多硫化物浓度随时间的变化(图2(a)和图2(b))。在红壤柱硫化过程中,在最初2 h内没有检出多硫化物,随后多硫化物浓度逐渐提高,在12 h时基本达到初始多硫化物入水浓度(50 mmol·L−1)。在褐土柱硫化过程中,多硫化物浓度随时间的变化与红壤柱类似,经过1.5 h后,开始检出多硫化物,并在12 h时达到平衡。
上述2个土柱的硫化过程中也观察到出水溶液的pH、氧化还原电位随时间的变化。对红壤柱,出水溶液pH变化从初始的5.6逐渐上升,12 h后达到12.7;出水溶液的氧化还原电位从初始的152.7 mV降至-582.5 mV。褐土柱出水溶液初始pH为7.3,经12 h硫化后,逐步升高至12.9;溶液氧化还原电位则从初始的136.4 mV逐渐下降至-595.8 mV。2种土壤硫化过程中pH和氧化还原电位的变化是由多硫化物所造成的。多硫化物是强碱性的物质,可导致出水溶液pH升高;同时由于其具有很强的还原特性,从而使得出水溶液氧化还原电位大大降低,并最终在柱体内建立有效的还原性区域。
2.2 硫化土柱对Cr(Ⅵ)的还原固定化
图3(a)和图3(b) 为红壤和褐土经硫化后还原固定化Cr(Ⅵ)的性能随时间变化的柱实验结果。由图3可知,在还原固定化过程中,柱体颜色、出水溶液的颜色、pH、氧化还原电位以及出水溶液中Cr(Ⅵ)和多硫化物的浓度均随时间发生变化。2种硫化土柱具有相似的变化趋势,但也存在一定的差异。
 图 3 出水溶液的pH、氧化还原电位、Cr(VI)浓度及
图 3 出水溶液的pH、氧化还原电位、Cr(VI)浓度及S2−x 浓度随时间的变化Figure 3. Changes of pH, oxidation-reduction potential, Cr(VI) andS2−x concentrations in the outlet solution with time当红壤柱和褐土柱还原固定化Cr(Ⅵ)时,柱体与出水液体颜色均随时间发生了变化。在柱实验过程中,随着对Cr(Ⅵ)的还原固定化,硫化红壤柱的颜色自下而上由深墨绿色逐渐变成红色,而其出水溶液颜色则由深墨绿色逐渐变淡,直至柱体还原能力消失,并最终变为淡黄色。硫化褐土柱在与Cr(Ⅵ)反应过程中,反应初始阶段柱体的颜色为绿色,随着反应时间的增加,其颜色自下而上逐渐变为黄褐色,而反应后出水颜色由初始的草绿色逐渐变为柱实验接近结束时的淡黄色。在2个硫化柱实验中,柱体中的绿色可能缘于硫化过程中所产生的亚铁与硫铁化合物。随着还原性物质的反应其颜色逐渐消失。淡黄色的出水接近入水Cr(Ⅵ)的本色。这主要是因为硫化过程中柱体内所产生的还原性物质随着与Cr(Ⅵ)反应,浓度逐渐降低;当还原性物质被彻底消耗后,入水和出水中Cr(Ⅵ)浓度接近,从而使得出水颜色呈淡黄色。
在硫化红壤柱和褐土柱还原固定化Cr(Ⅵ)的过程中,也观察到了其出水溶液pH和氧化还原电位类似的变化情况(图3(a)和图3(b))。对硫化红壤柱,出水溶液初始pH为12.6,之后随反应的进行,pH逐步下降,最终达到8.9;出水溶液氧化还原电位从-574.9 mV逐渐上升至117 mV。硫化褐土柱的出水溶液pH从12.9下降至9.4,氧化还原电位则从-571.9 mV逐渐上升至38.5 mV。柱实验中pH和氧化还原电位的变化是由于多硫化物和Cr(Ⅵ)造成的。多硫化物是强碱性的物质,随着与Cr(Ⅵ)的反应,其浓度逐渐降低,造成了pH和氧化还原电位的上述变化。
实验中观察到Cr(Ⅵ)浓度随时间发生变化。向硫化红壤柱持续通入含10 mg·L−1Cr(Ⅵ)地下水27 h(约38PV)后,土柱达到穿透点(0.1 mg·L−1)。由于已穿透的土柱仍然具有一定的还原能力,因此,可以进一步还原固定化Cr(Ⅵ),但其还原固定化能力的降低,导致出水中Cr(Ⅵ)的浓度逐渐升高,并最终在155 h (约218PV)时达到平衡。通过积分可得,硫化红壤柱在整个实验过程中共去除1.21 mmol Cr(Ⅵ),由于Cr(Ⅵ)被还原固定化为Cr(Ⅲ),即反应过程中需净利用3.63 mmol电子。硫化褐土柱也经历了类似的过程,在通入10 mg·L−1 Cr(Ⅵ) 15 h (约22PV)后,土柱达到穿透点。之后该土柱仍能继续还原Cr(Ⅵ),并在反应96 h (约138PV)时丧失还原能力。同样由积分可知,硫化褐土柱在整个反应过程中共去除0.77 mmol Cr(Ⅵ),即2.31 mmol净利用电子量。柱实验中,土柱累积去除Cr(Ⅵ)的量可根据式(1)计算。
S=12n0∑i=1[(C0−Ci+1)+(C0−Ci)]⋅q⋅(ti+1−ti) (1) 式中:Ci为取样时间ti时出水中Cr(Ⅵ)浓度,mg·L−1;C0为入水中Cr(Ⅵ)浓度,mg·L−1;q为入水流速,取值1.15 mL·min−1;n0为取样数据点数。
在2个硫化土柱还原Cr(Ⅵ)过程中,反应初期在出水中还检测到多硫化物存在。向硫化红壤通入Cr(Ⅵ)溶液,出水中多硫化物的浓度在2 h内由初始的50 mmol·L−1迅速降低到约10 mmol·L−1,并最终在10 h后接近0。对褐土柱来说,出水中多硫化物浓度变化的趋势与上述红壤中观察到的变化趋势类似:多硫化物浓度在经历了2 h快速下降后,并在反应7.5 h后,浓度逐渐接近0。经过硫化的土柱孔隙中仍存在未反应的多硫化物,这些多硫化物在后续通入Cr(Ⅵ)溶液时被取替而流出,因此造成了多硫化物仍出现在出水中。考虑到扣除流出的这部分多硫化物后,是实际消耗的多硫化物,即为硫化土柱所持有的净还原能力,因此,硫化红壤柱所持有的净还原能力相当于7.89 mmol多硫化物。由于
S2−x 在氧化过程中的产物以S0为主,而1 molS2−x 在氧化还原反应中能提供2 mol电子,因此,硫化红壤所持有的净还原能力相当于15.78 mmol电子。对褐土柱来讲,柱体所持有的净还原能力相当于4.65 mmol多硫化物或9.3 mmol电子。综合以上实验结果,可以看出,由多硫化物在柱体内所形成的理论还原能力与还原Cr(Ⅵ)中实际消耗的还原能力存在偏差。如果定义柱实验过程中的电子利用效率为Cr(Ⅵ)还原固定化所需理论电子数与柱体所持有总还原能力对应的电子数之比,基于柱实验还原固定化Cr(Ⅵ)的结果可知,硫化后的红壤和褐土的电子利用效率分别为23.0%和24.8%,相当于去除等当量的Cr(Ⅵ)所需理论还原能力的4.35倍和4.03倍。柱实验与理论值的偏差可能是由于土壤复杂性所造成的。因此,以多硫化物为还原剂,采用ISRM修复技术还原固定化Cr(Ⅵ)时,需要加入过量还原试剂。基于对红壤柱和褐土柱实验结果,可以看出,柱实验中所用的多硫化物是所需理论值的4~5倍。这一结果对受Cr(Ⅵ)污染的土壤-地下水修复具有一定的实际意义。
3. 结论
1)经硫化处理的2种土壤均具有还原固定化Cr(Ⅵ)的能力,但其还原容量不同。在实验条件下,向硫化土柱中通入10 mg·L−1的Cr(Ⅵ)溶液,红壤柱和褐土柱分别在38PV和22PV穿透(基于GB/T 14848-2017,穿透浓度设为0.1 mg·L−1)。2种土柱在穿透后虽然其还原能力降低但仍能持续的还原固定化Cr(Ⅵ),并最终在218PV和138PV左右丧失对Cr(Ⅵ)的还原能力。
2)多硫化物对红壤和褐土的硫化会使孔隙水的pH和氧化还原电位发生变化。在经硫化处理后,红壤柱和褐土柱的出水pH分别升高至12.7和12.9,而对应的氧化还原电位则逐渐降低至-582.5 mV和-595.8 mV,这表明多硫化物在土壤中已建立有效的还原区域。向2种硫化土柱中通入Cr(Ⅵ)溶液会降低出水的pH,同时伴随氧化还原电位的升高:反应结束时,红壤和褐土系统的pH分别为8.9和9.4,对应的氧化还原电位分别为117 mV和38.5 mV。
3)经多硫化物硫化后的红壤和褐土在还原Cr(Ⅵ)过程中实际电子利用效率分别为23.0%和24.8%,即实际所需多硫化物的量是理论值的4~5倍。考虑到污染现场地下介质的非均质性和复杂性,在实际Cr(Ⅵ)修复中多硫化物的用量可能会更高。
-
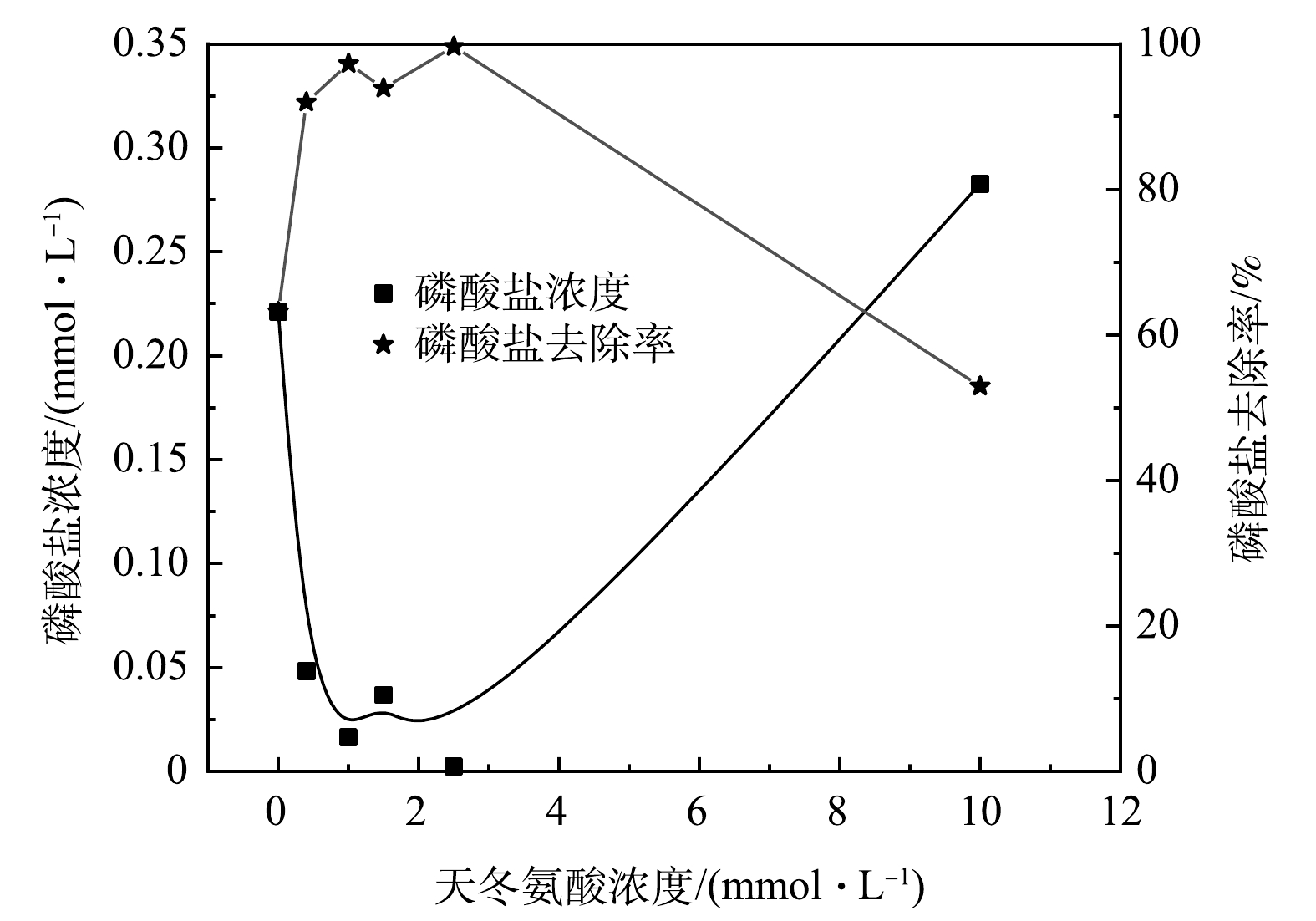
图 2 不同浓度天冬氨酸添加下磷酸盐的残余量
Figure 2. Residual amount of phosphate at different concentration of aspartic acid

图 5 1.5 mmol·L-1天冬氨酸浓度时絮体的TEM图
Figure 5. TEM images of flocs at 1.5 mmol·L-1 aspartic acid
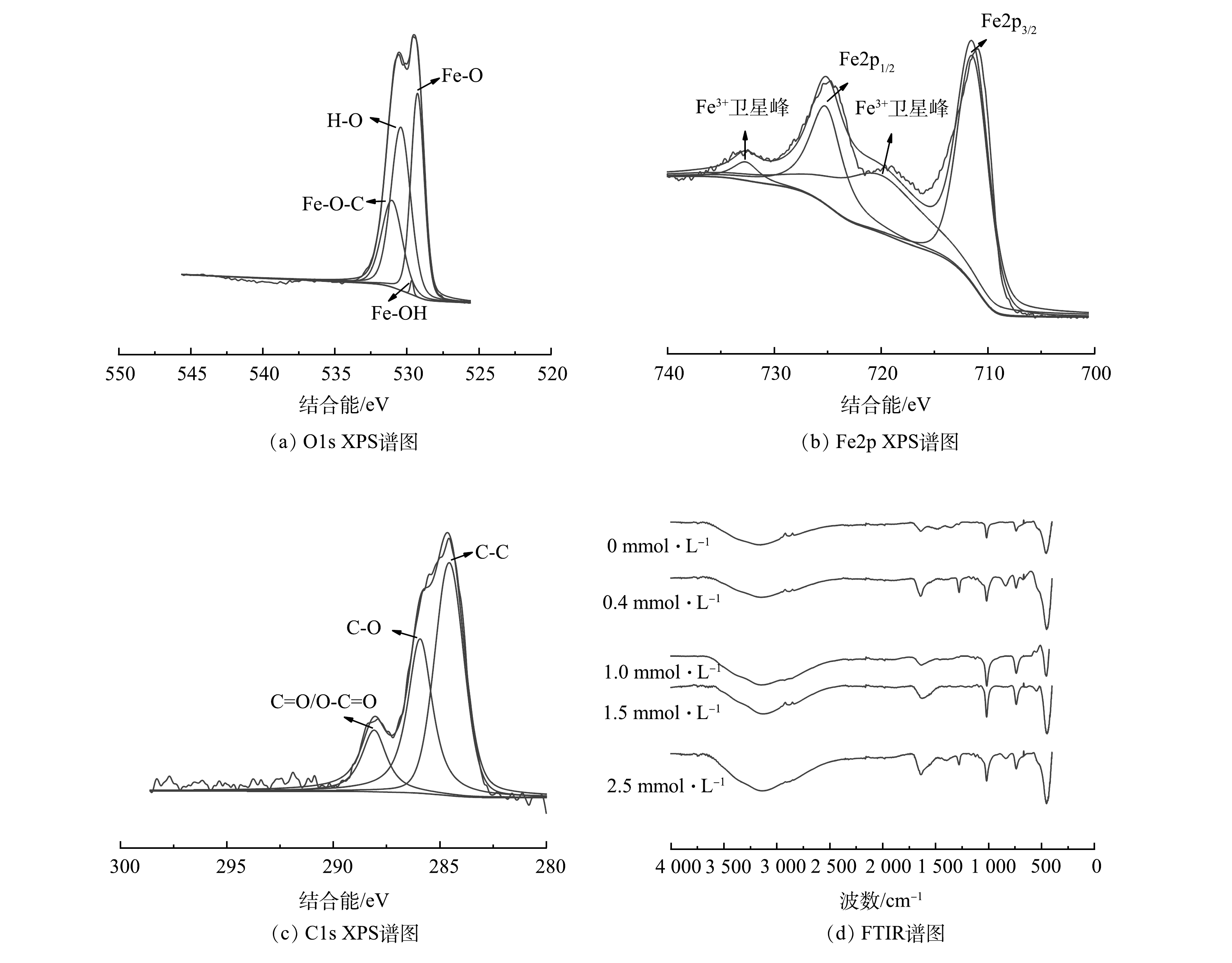
图 6 添加1.5 mmol·L-1天冬氨酸时絮体的表面XPS和FTIR图谱
Figure 6. FTIR and XPS spectra of floc surface at 1.5 mmol·L-1 aspartic acid
-
[1] SILLANPAA M, NCIBI M C, MATILAINEN A, et al. Removal of natural organic matter in drinking water treatment by coagulation: A comprehensive review[J]. Chemosphere, 2018, 190: 54-71. doi: 10.1016/j.chemosphere.2017.09.113 [2] XU W, GAO B, YUE Q, et al. Effect of shear force and solution pH on flocs breakage and re-growth formed by nano-Al(13) polymer[J]. Water Research, 2010, 44(6): 1893-1899. doi: 10.1016/j.watres.2009.11.029 [3] YU W, GREGORY J, CAMPOS L C. Breakage and re-growth of flocs: Effect of additional doses of coagulant species[J]. Water Research, 2011, 45(20): 6718-6724. doi: 10.1016/j.watres.2011.10.016 [4] YU W, GREGORY J, CAMPOS L C, et al. Dependence of floc properties on coagulant type, dosing mode and nature of particles[J]. Water Research, 2015, 68: 119-126. doi: 10.1016/j.watres.2014.09.045 [5] WU M, YUAN J, WU H, et al. Ultrathin nanofiltration membrane with polydopamine-covalent organic framework interlayer for enhanced permeability and structural stability[J]. Journal of Membrane Science, 2019, 576: 131-141. doi: 10.1016/j.memsci.2019.01.040 [6] HSU P H. Comparison of iron(III) and aluminum in precipitation of phosphate from solution[J]. Water Research, 1976: 903-907. [7] SU Z, LIU T, YU W, et al. Coagulation of surface water: Observations on the significance of biopolymers[J]. Water Research, 2017, 126: 144-152. doi: 10.1016/j.watres.2017.09.022 [8] YU W, XU L, LEI K, et al. Effect of crystallization of settled aluminum hydroxide precipitate on "dissolved Al"[J]. Water Research, 2018, 143: 346-354. doi: 10.1016/j.watres.2018.06.063 [9] BOLAND D D, COLLINS R N, MILLER C J, et al. Effect of solution and solid-phase conditions on the Fe(II)- accelerated transformation of ferrihydrite to lepidocrocite and goethite[J]. Environmental Science & Technology, 2014, 48(10): 5477-5485. [10] SHU Z, LIU L, TAN W, et al. Solar irradiation induced transformation of ferrihydrite in the presence of aqueous Fe2[J]. Environmental Science & Technology, 2019, 53(15): 8854-8861. [11] MIRABELLO G, IANIRO A, BOMANS P H H, et al. Crystallization by particle attachment is a colloidal assembly process[J]. Nature Materials, 2020, 19(4): 391-396. doi: 10.1038/s41563-019-0511-4 [12] OSTWALD W. Studien über die bildung und umwandlung fester Körper: 1. Abhandlung: Übersättigung und Überkaltung[J]. Zeitschrift für Physikalische Chemie, 1897, 22U(1): 289-330. [13] ZHANG X, SHEN Z, LIU J, et al. Direction-specific interaction forces underlying zinc oxide crystal growth by oriented attachment[J]. Nature Communications, 2017, 8(1): 835. doi: 10.1038/s41467-017-00844-6 [14] ZHU C, LIANG S, SONG E, et al. In-situ liquid cell transmission electron microscopy investigation on oriented attachment of gold nanoparticles[J]. Nature Communications, 2018, 9(1): 421. doi: 10.1038/s41467-018-02925-6 [15] XING B, GRAHAM N, YU W. Transformation of siderite to goethite by humic acid in the natural environment[J]. Communications Chemistry, 2020, 3(1): 38. doi: 10.1038/s42004-020-0284-3 [16] GUAN X H, CHEN G H, SHANG C. Combining kinetic investigation with surface spectroscopic examination to study the role of aromatic carboxyl groups in NOM adsorption by aluminum hydroxide[J]. Journal of Colloid and Interface Science, 2006, 301(2): 419-427. doi: 10.1016/j.jcis.2006.05.031 [17] SHENG A, LIU F, XIE N, et al. Impact of proteins on aggregation kinetics and adsorption ability of hematite nanoparticles in aqueous dispersions[J]. Environmental Science & Technology, 2016, 50(5): 2228-2235. [18] YU W Z, GREGORY J, CAMPOS L, et al. The role of mixing conditions on floc growth, breakage and re- growth[J]. Chemical Engineering Journal, 2011, 171(2): 425-430. doi: 10.1016/j.cej.2011.03.098 [19] YU W Z, GREGORY J, GRAHAM N. Regrowth of broken hydroxide flocs: Effect of added fluoride[J]. Environmental Science & Technology, 2016, 50(4): 1828-1833. [20] XING B, OUYANG M, GRAHAM N, et al. Enhancement of phosphate adsorption during mineral transformation of natural siderite induced by humic acid: Mechanism and application[J]. Chemical Engineering Journal, 2020, 393: 124730. doi: 10.1016/j.cej.2020.124730 [21] THOMASARRIGO L K, BYRNE J M, KAPPLER A, et al. Impact of organic matter on iron(II)-catalyzed mineral transformations in ferrihydrite-organic matter coprecipitates[J]. Environmental Science & Technology, 2018, 52(21): 12316-12326. [22] CHEN C, KUKKADAPU R, SPARKS D L. Influence of coprecipitated organic matter on Fe2+(aq)-catalyzed transformation of ferrihydrite: Implications for carbon dynamics[J]. Environmental Science & Technology, 2015, 49(18): 10927-10936. [23] CHEN C, DYNES J J, WANG J, et al. Properties of Fe-organic matter associations via coprecipitation versus adsorption[J]. Environmental Science & Technology, 2014, 48(23): 13751-13759. [24] WECKLER H D L. Lattice vibration spectra. Part XCV. Infrared spectroscopic studies on the iron oxide hydroxides goethite (α), akaganéite (β), lepidocrocite (γ), and feroxyhite (δ)[J]. European Journal of Solid State and Inorganic Chemistry, 1998, 35(8-9): 531-544. doi: 10.1016/S0992-4361(99)80017-4 [25] LI X, GRAHAM N J D, DENG W, et al. The formation of planar crystalline flocs of γ-FeOOH in Fe(II) coagulation and the influence of humic acid[J]. Water Research, 2020, 185: 116250. doi: 10.1016/j.watres.2020.116250 [26] Bio-inspired fabrication of hierarchical FeOOH nanostructure array films at the air-water interface, their hydrophobicity and application for water treatment[J]. ACS nano, 2013, 72: 1368-1378. -

 点击查看大图
点击查看大图

计量
- 文章访问数: 4111
- HTML全文浏览数: 4111
- PDF下载数: 99
- 施引文献: 0
