

 下载:
下载:





-
近年来,纺织印染废水处理已成为制约我国纺织印染行业可持续发展的重要问题。在地方和行业排放标准提高的背景下,采用传统水处理工艺如生物法、絮凝沉淀法和吸附法单独处理上述废水时往往难以达标,而化学氧化法、光催化氧化法和电化学法又由于运行成本的高昂未能在利润微薄的印染企业得以推广。化学氧化法包括臭氧氧化法、芬顿法和高温深度氧化法。较之其他的化学氧化法,芬顿法在目前难降解印染废水处理中应用较多,但其也存在铁泥处理、大量酸碱调节pH、亚铁离子催化作用慢等问题[1-4]。次氯酸钠具有强氧化性,且价格低廉(1t含10%~11%有效氯的液体次氯酸钠的价格为600元)。但次氯酸钠在温度较高或者在日光照射的条件下,容易发生分解反应,生成氯酸钠、氯化钠和氯化氢气体等,因而大大降低了其强氧化性的利用率。如何提高次氯酸钠氧化性能的利用率对拓宽其在水处理中的应用至关重要。
次氯酸钠的氧化性在镍、铁、锰等过渡金属氧化物/氢氧化物的作用下可大大增强[5-6]。其中,镍基催化剂由于具有催化活性高、分散性好和价格低廉的优点而备受关注[7-9]。有学者[10-11]采用NaClO/Ni2O3催化体系处理印染废水,取得了较好的效果,但催化剂Ni2O3在应用过程中的转化、溶解和处理后出水中Ni2+溶出量方面研究的缺失限制了该催化氧化工艺的应用及推广。此外,市售Ni2O3为黑色粉末,若直接用于水处理的话,很容易流失,从而导致金属镍的二次污染。有科研人员[7-9]在400~500 ℃高温焙烧制取镍基催化剂,用于促进次氯酸钠的分解。本课题组的预研结果表明,用高温焙烧法制取的颗粒型催化剂不能有效地促进次氯酸钠的分解。
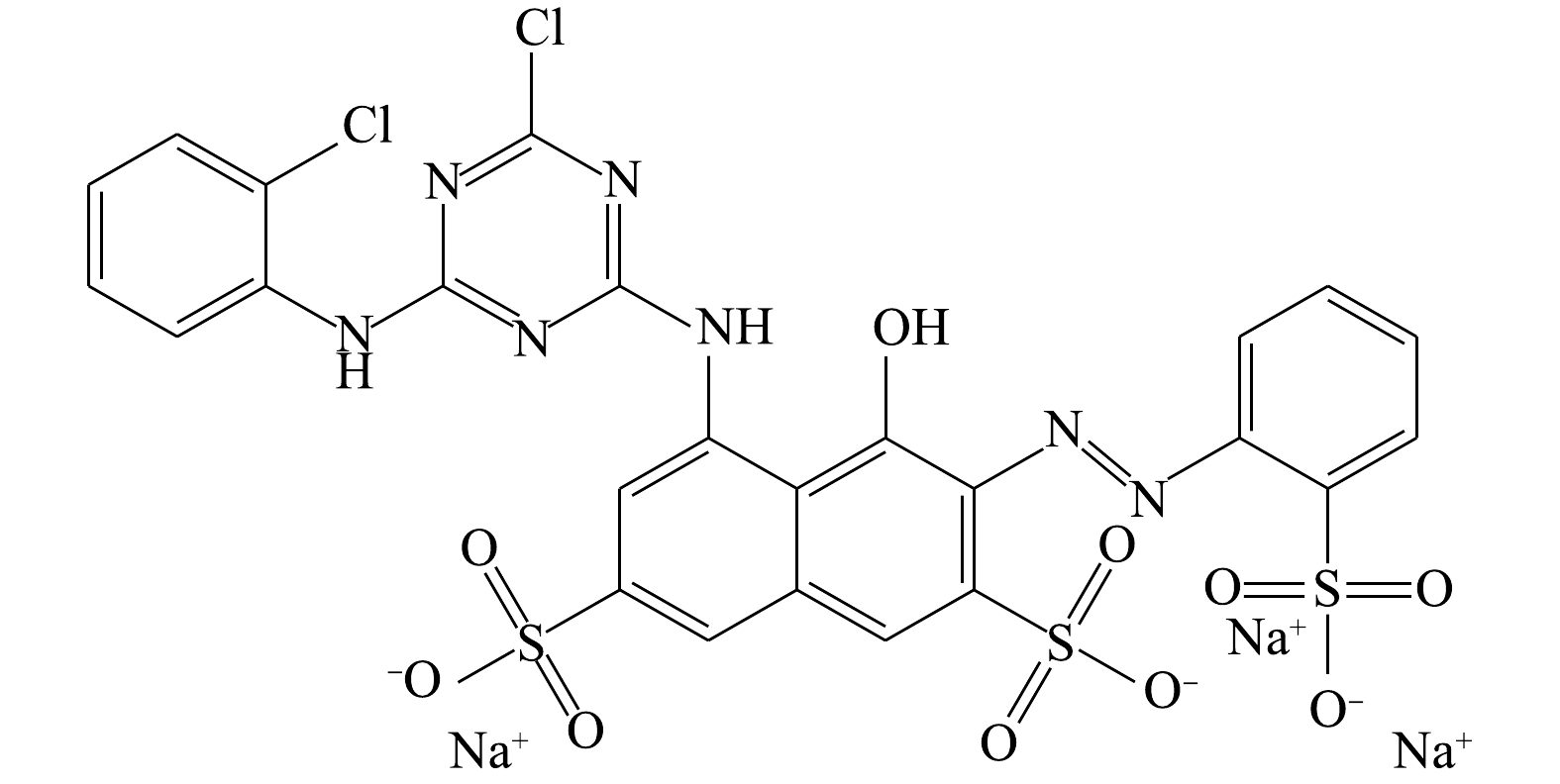
活性艳红K-2BP分子式为C25H14N7Na3O10S3Cl2,相对分子质量为808.48,最大吸收波长为534.5 nm。K-2BP分子结构包含1个偶氮基,一氯均三嗪基及2个苯环和1个萘环。苯环和萘环上有3个磺酸基(图1)。这些芳香环上的共轭链使染料显色,也使其具备了高水溶性、在自然条件下难降解的特性。一般来说,染料分子结构中共轭链越长,颜色越深。采用生物法降解这类染料,一般需要厌氧/好氧组合工艺才能获得良好的处理效果。顾梦琪等[12]采用水解酸化/AO组合工艺处理活性艳红X-3B的印染废水,取得了良好的效果。总的来说,如何低成本、高效率地处理这类高色度及难降解的废水一直是水处理领域的一项难题[13-14]。
本文旨在采用简单易行的氧化铝小球浸渍法制备一种使用方便、催化效果好、不会流失、不产生二次Ni2+污染的镍基催化剂NiOx(OH)y,并采用NaClO和该催化剂组成的体系降解模拟印染废水中的活性艳红K-2BP,考察了在不同反应条件下该体系对活性艳红K-2BP的处理效果和处理后出水中Ni2+的溶出情况,以期为开发一种性价比较高的水处理催化氧化新工艺提供参考。
全文HTML
-
催化剂载体选用洁之源水处理材料厂的γ-Al2O3小球。该小球表面具有丰富的毛细孔,这些孔道具有较强的吸附能力,能有效吸附溶液及其中的分子和离子。γ-Al2O3参数如下:直径为2~3 mm、氧化铝含量≥92%、孔容为0.35 mL·g−1、比表面积S≥300 m2·g−1。γ-Al2O3俗称刚玉,耐酸碱。
在本研究中,选用过硫酸钠作为制备镍基催化剂的氧化剂。和其他氧化剂比较,过硫酸钠具有反应pH范围宽、溶解度好、活化后能够同时产生
SO−4 ·和·OH的优点。相比于·OH (E0=1.8~2.7 V),SO−4 ·具有更高的氧化电位(E0=2.5~3.1 V)和更长的半衰期,并且过硫酸钠在碱性条件下也可以产生具有强氧化性的SO−4 ·[15-16]。Ni2+与Na2S2O8在碱性条件下反应生成NiOOH与Ni(OH)2的混合物,采用化学式NiOx(OH)y表示该混合物。将负载有Na2S2O8-NaOH混合液的γ-Al2O3小球浸泡在含有Ni2+的溶液里发生反应,在γ-Al2O3表面形成一层NiOx(OH)y沉淀,从而得到NiOx(OH)y/Al2O3催化剂小球。在制备催化剂之前,先用蒸馏水洗净载体γ-Al2O3小球表面的粉尘和其中的木屑,然后再放入箱式电阻炉中于200 ℃下焙烧2 h,从炉中取出,待冷却至室温后称量使用。
浸渍步骤共分为以下3个步骤:按一定比例和浓度配置Na2S2O8-NaOH混合液,称取一定量的γ-Al2O3小球,与该混合液一起放入100 mL容量的锥形瓶中,室温下在振荡器上以100 r·min−1振荡2 h,将得到的Na2S2O8-NaOH/Al2O3小球滤出,烘干;配置一定浓度的Ni(NO3)2溶液,再将烘干后的小球和Ni(NO3)2溶液放入锥形瓶中,室温下在振荡器上以100 r·min−1的频率振荡2 h,将制得的NiOx(OH)y/Al2O3小球滤出,烘干;将制得的NiOx(OH)y/Al2O3小球与配制的Na2S2O8-NaOH混合液置于锥形瓶中,室温下在振荡器上以100 r·min−1振荡2 h,将NiOx(OH)y/Al2O3小球滤出,用蒸馏水清洗3次。最后,将洗净后的NiOx(OH)y/Al2O3置于马弗炉中于100 ℃下焙烧2 h,制得次氯酸钠催化氧化实验所需的催化剂样品。
所制得的NiOx(OH)y/Al2O3催化剂呈蛋壳型,其表面裹着一层黑绿色的NiOx(OH)y膜,厚度约为0.1 mm(图2)。大量Ni2+与碱反应生成Ni(OH)2,生成的部分Ni(OH)2与过硫酸钠反应生成NiOOH;催化剂的内部仍为白色的Al2O3,Ni2+没有渗透到小球内部,这是因为小球表面生成的黑绿色的NiOx(OH)y膜阻碍了Ni2+的渗透。
当化学式NiOx(OH)y中的x=1、y=1时,Ni2+在碱性溶液中被Na2S2O8氧化的过程如式(1)所示,式(1)为式(2)和式(3)的总和。
由式(1)~式(3)可知,过硫酸钠与镍(Ⅱ)完全反应后,产物包括Ni(OH)2、NiOOH、水和
SO2−4 。其中,SO2−4 能很容易地被冲洗掉。由式(1)可知,碱性条件促进Ni2+的氧化。在羟基氧化镍NiOOH的存在下,次氯酸钠的分解反应可以看作是式(4)和式(5)的总和。产生的氧分子能被物理吸附在镍基催化层上,捕获电子后转化为化学吸附氧
O−2 和O−以及原子氧O(式(7)~式(8))。物理吸附的氧分子也可以直接分解成原子氧[17](式(9)) 。式中:O2(g)为气态氧;S为表面吸附位置;O2为表面物理吸附的氧;*和e−代表导带底部空位和导带电子;∆G为反应能。反应产生的原子氧O非常活跃,可以快速氧化和分解有机物[18]。羟基氧化镍可以长时间用作催化剂,因为它可以按照式(4)和式(5)进行再生和再利用。
-
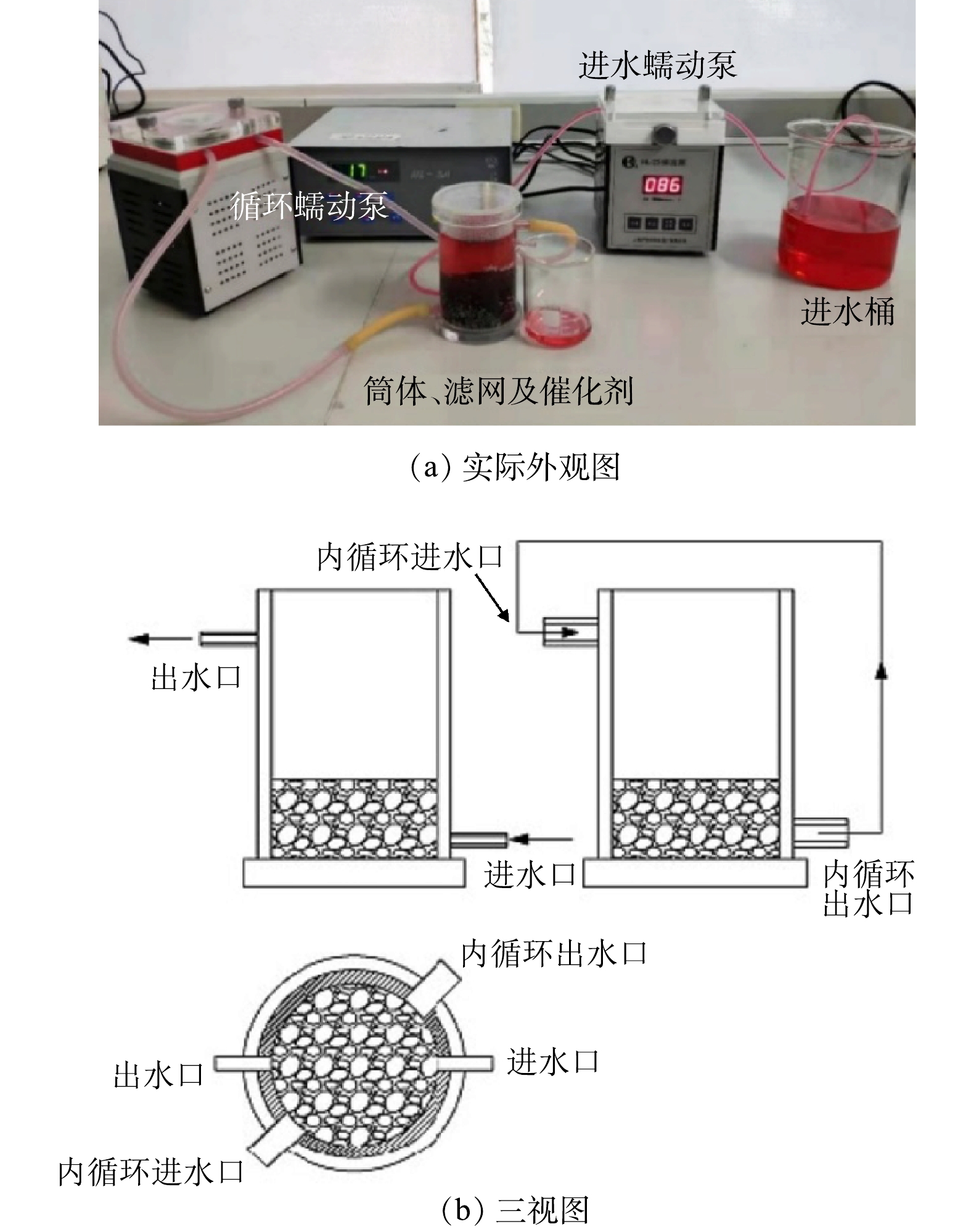
本研究所用的连续流实验装置由反应器、进水泵、循环泵以及进水桶等组成,实物图及平面三视图见图3。反应器(Φ为6 cm,H为10 cm)材质为有机玻璃,在反应器的内壁靠近出水口贴着一层尼龙滤网(孔径为2 mm),以防催化剂小球的流失。模拟印染废水下进上出,内循环流上进下出,以使反应器内水质混合均匀,内循环流的流速约为进水流速的5倍。每天运行360 min或者540 min,总共运行了20 d。
-
过硫酸钠(99%)、氢氧化钠(99%)、六水合硝酸镍(99%)、浓硫酸(98%)、呋喃甲醇和叔丁醇均为分析纯,购自国药集团化学试剂有限公司;次氯酸钠溶液含有效氯≥10%。
ZYL便携式余氯分析仪,杭州齐威仪器有限公司;SD90740镍离子浓度测定仪,上海昕瑞仪器仪表有限公司;MIT-3F型COD检测仪,常州三丰仪器科技有限公司;TOC-VCPH分析仪,日本岛津;723N可见光分光光度计,上海佑科仪器仪表有限公司;EscaLab 250Xi型X射线光电子能谱仪,美国Thermo Fisher,Al-Kα射线单色源;扫描电镜SEM, 美国FEI Inspect F50。
-
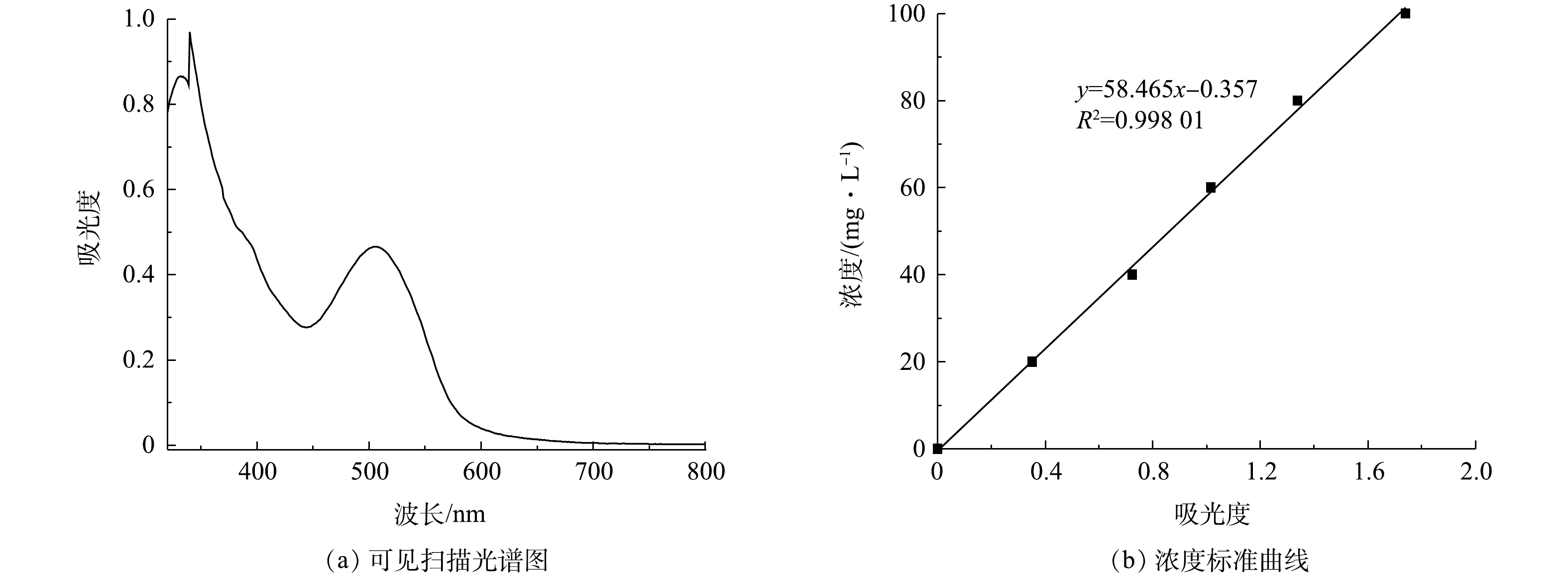
配置一定浓度的活性艳红K-2BP溶液,加入一定量的次氯酸钠溶液,充分搅拌,调节pH至所需值,得到印染废水/次氯酸钠混合液。取上述混合液50 mL置入100 mL锥形瓶内,称取一定量的催化剂样品快速放入上述溶液中。室温下在振荡器上以100 r·min−1的速度振荡120 min。分别在第10、20、30、50、70、90、120 min时取3 mL左右水样,测其吸光度,结果如图4所示,利用标准曲线法检测染料的浓度,按式(10)计算其脱色率。测好吸光度后将染料溶液倒回锥形瓶中继续反应。
式中:R为脱色率;C0和Ct分别为染料的初始浓度和t时刻的浓度,mg·L−1。
-
配置一定浓度的活性艳红K-2BP溶液,加入一定量的次氯酸钠溶液,充分搅拌,调节pH至所需值,得到印染废水/次氯酸钠混合液。在2个相同浓度的混合液中分别加入一定量的自由基清除剂叔丁醇和呋喃甲醇,与制备的催化剂样品一起放入100 mL锥形瓶内,室温下在振荡器上以100 r·min−1的速度振荡反应,检测染料的脱色率变化。
1.1. 催化剂制备
1.2. 连续流实验
1.3. 试剂及测试仪器
1.4. 染料脱色率分析
1.5. 自由基清除剂实验
-
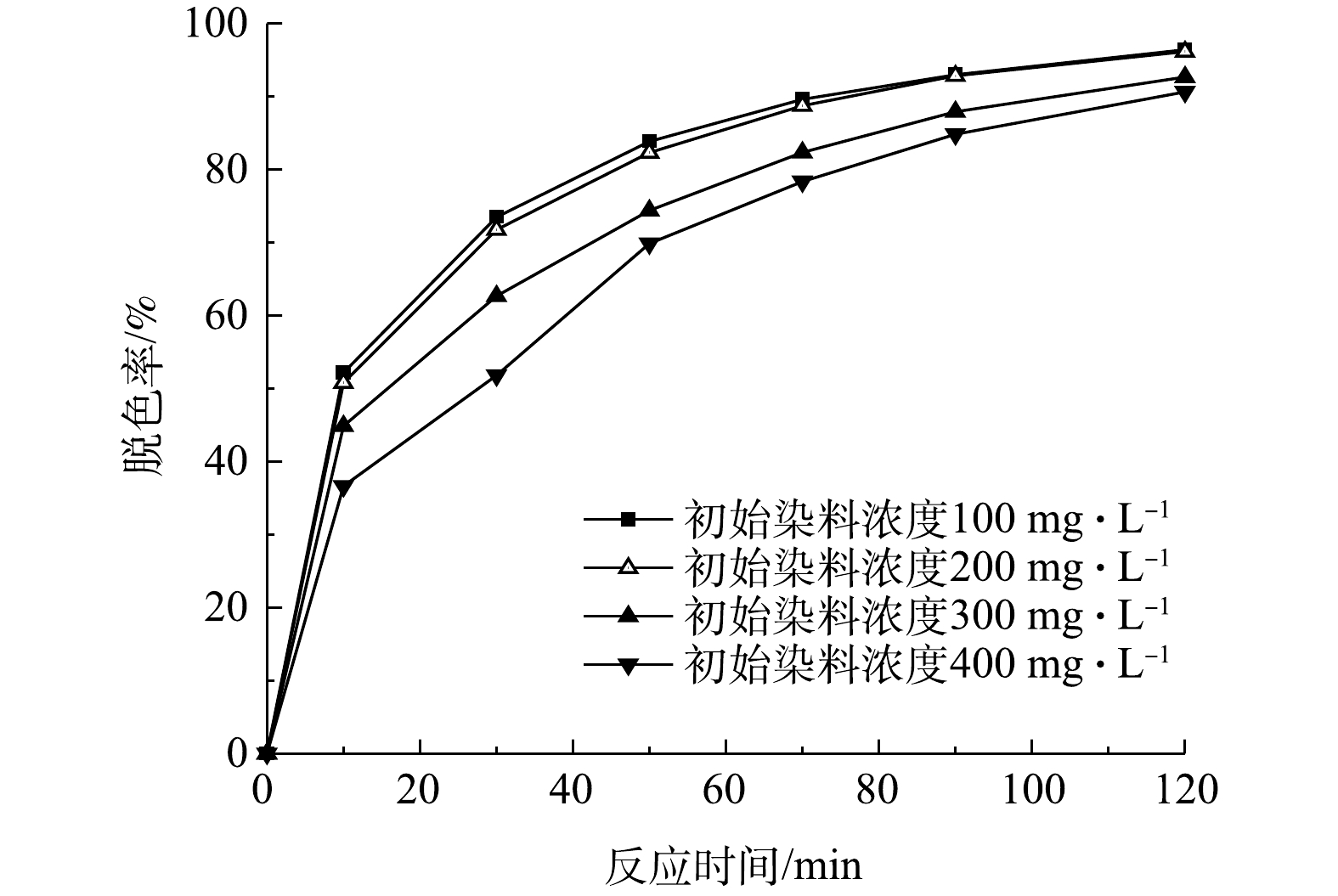
检测了当初始pH为9、初始有效氯浓度为120 mg·L−1、催化剂投加量为240 g·L−1、初始染料浓度为100、200、300、400 mg·L−1时,不同反应时间下的脱色率,结果如图5所示。由图5可以看出,经过120 min的处理,4种浓度的印染废水的脱色率均能达到90%以上。当初始染料浓度为100 mg·L−1和200 mg·L−1时,脱色率的值很接近,在反应50 min时脱色率达到80%以上。在处理初始浓度为400 mg·L−1的染料废水时,需要把处理时间延长到90 min以上才能达到80%以上的脱色率。
由表1可知,活性艳红K-2BP在不同初始浓度下的脱色降解过程都符合拟一级动力学方程。从200 mg·L−1的初始浓度开始,反应速率常数随着初始染料浓度的增加而降低,这是由于在4个体系中具有等量的有效氯和镍基催化剂,由此产生等量的活性氧。染料的分解受到活性氧产量的限制,其浓度的增加使反应速率变慢。在200 mg·L−1以下时没有出现这种现象。当染料浓度不高时,系统中产生的活性氧足以使偶氮键断裂,因此,初始浓度改变时速率常数变化不大。
-
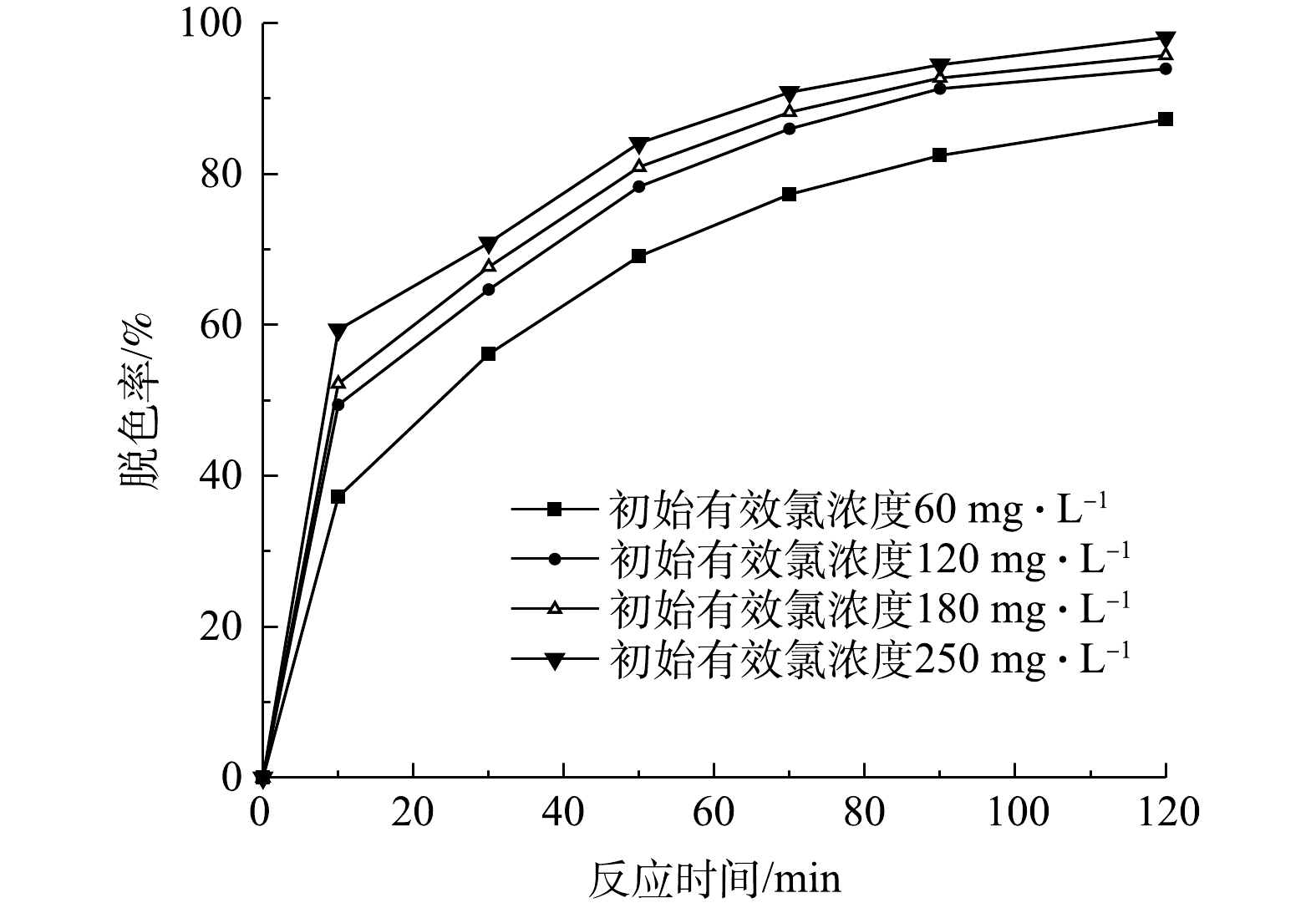
检测当初始pH为9、活性艳红K-2BP的初始浓度为200 mg·L−1、催化剂投加量为240 g·L−1、初始有效氯浓度分别为60、120、180、250 mg·L−1时,在不同反应时间下K-2BP的脱色率,结果如图6所示。由图6可以看出,当初始有效氯浓度为180 mg·L−1和250 mg·L−1时,K-2BP的脱色率在50 min时均达到80%以上,而初始有效氯浓度为60 mg·L−1的水样在90 min时脱色率才能达到80%。
由表2可见,在不同的初始有效氯浓度下,活性艳红K-2BP脱色反应符合拟一级动力学方程。其中,反应速率常数随着初始有效氯浓度的增加而增大,这是由于次氯酸钠催化分解后产生的活性氧能与染料分子进行反应,增大初始有效氯浓度可以促进活性氧产量的提升,从而加快活性艳红K-2BP溶液的脱色反应,拟一级反应速率常数也随之增大。此外,反应速率增加的幅度随着有效氯浓度的增加逐渐变小,这是因为随着活性氧数量的增加,易降解的共轭发色基团逐渐分解,模拟废水颜色变浅,但染料并没有完全矿化,活性氧会继续与其他的如萘环、均三嗪等部分难降解基团反应。这些基团本身不显色,但会随着氧化反应的深入而逐步开环分解为更小的分子结构[19]。此外,活性氧的数量和有效氯浓度之间不是简单的线性正比例关系,当催化剂的量一定时,活性氧的产量随着有效氯浓度的增加而增加,增加的幅度减小,并在一定的有效氯浓度下达到最大值,继续增加有效氯浓度也不会增加活性氧的数量。所以,从脱色率上看,反应速率增加的幅度变小了。
-
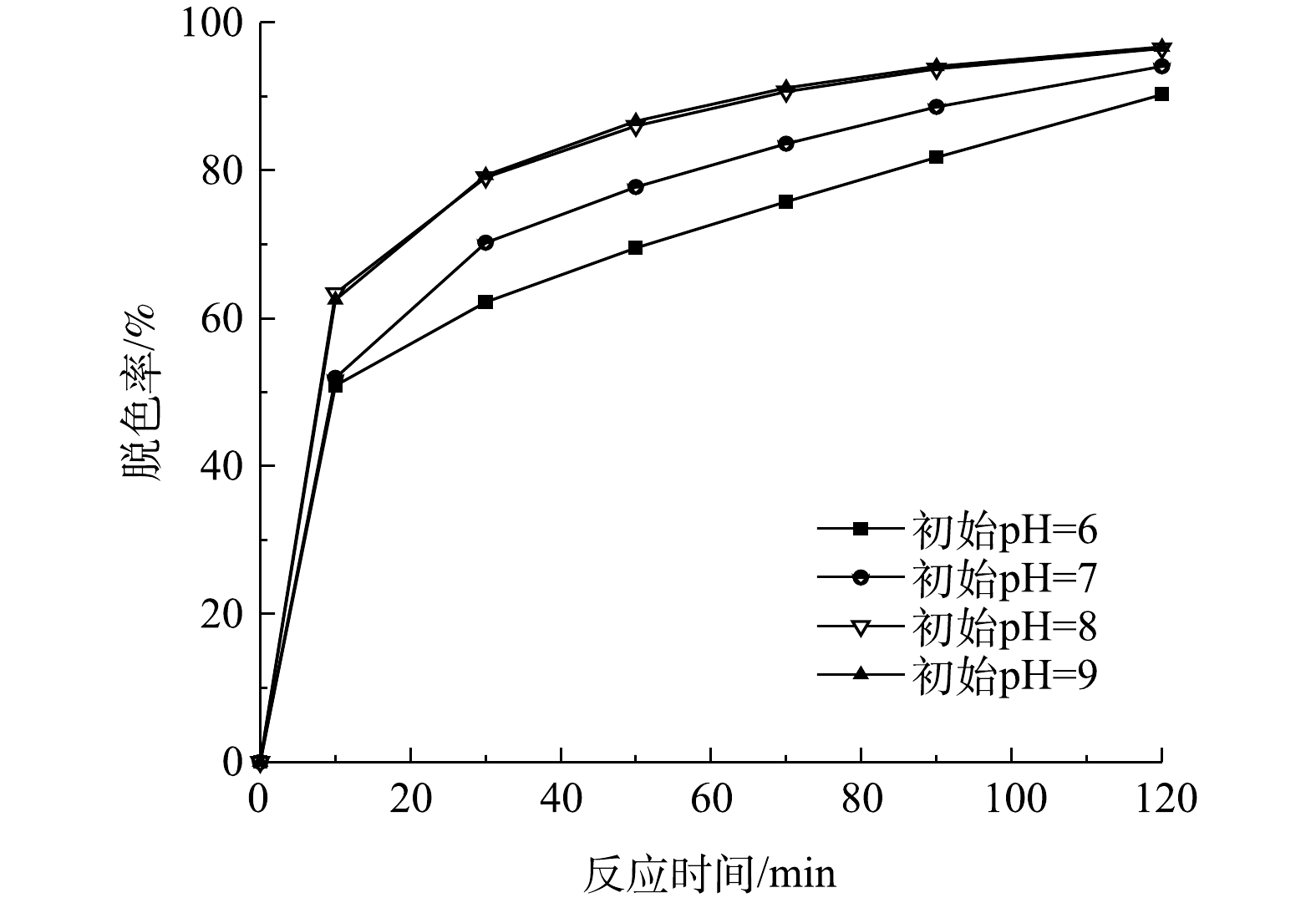
检测当初始有效氯浓度为120 mg·L−1、初始染料浓度为200 mg·L−1、催化剂投加量为240 g·L−1、初始pH为6、7、8、9时,不同反应时间下的脱色率,结果如图7所示。由图7可以看出,当初始pH为8和9时,2个溶液体系中的脱色效果基本相同,当反应50 min时,脱色率已经达到86%以上。而初始pH为7时的脱色效果次于初始pH为8和9时的,初始pH为6时脱色效果最差,当反应90 min时此体系中的脱色率才达到80%以上。
由表3可见,不同初始pH时次氯酸钠催化降解活性艳红K-2BP的过程符合拟一级动力学方程,而且反应速率常数随着初始pH的增加而增加,这是因为在染料降解过程中,溶液pH会持续性降低。ZENG等[19]发现活性艳红K-2BP经次氯酸钠氧化降解后,pH由7.5左右降至4.65左右。在反应过程中染料分子的酚基和HClO的电离过程中会释放出氢离子(式(11)和式(12));此外,在染料降解过程中可能产生芳香酸和脂肪酸,也会降低溶液pH。因此,弱碱性的初始pH有利于中和反应过程产生的氢离子,从而减小pH的降低幅度。本课题组的前期研究结果[20]表明,采用该类催化剂分解次氯酸钠,当溶液的初始pH为8和9时,次氯酸钠的转化率最高,产生最多的活性氧,从而有利于染料的脱色降解效果。
-
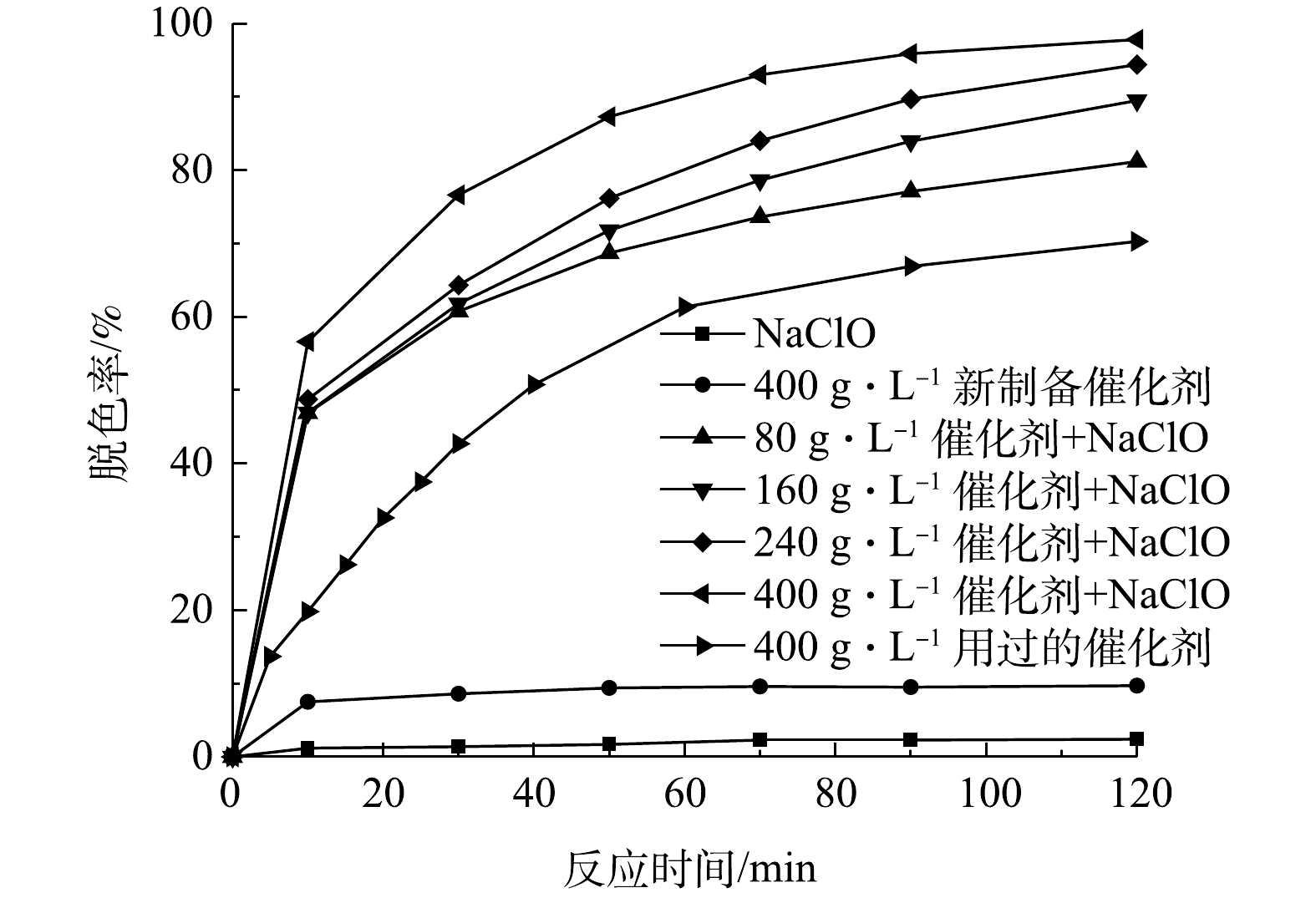
测定当初始有效氯浓度为120 mg·L−1、初始染料浓度为200 mg·L−1、初始pH为9、催化剂投加量为0、80、160、240、400 g·L−1时,不同反应时间下的脱色率,结果如图8所示。由图8可以看出,催化剂投加量的增加显著改善了活性艳红K-2BP的脱色效果。无催化剂时,次氯酸钠基本不能降解活性艳红K-2BP,该染料化学性质相当稳定;催化剂投加量为80 g·L−1溶液时,脱色率直到90 min时仍未能达到80%;当投加量增加至400 g·L−1时,脱色率在90 min时能达到96%。
由表4可以看出,在80~400 g·L−1的催化剂投加量下,脱色反应符合拟一级动力学方程。催化剂投加量变大增加了催化剂表面活性位点的数量,提升了其对有效氯的吸附和分解速率,染料和中间产物对催化剂表面的位点的竞争吸附减少,因此,提高了染料降解反应速率常数,改善了染料的脱色效果。在不使用氧化剂,单独使用400 g·L−1新制备的催化剂处理废水120 min后,脱色率仅为9.7%,而且随着反应时间的延长,脱色率变化幅度很小,说明这部分脱色率是由吸附主导的。单独使用400 g·L−1批量实验用过6次的(每次使用时间为120 min)催化剂处理废水,发现脱色率随着反应时间的延长增加,其增速比(NaClO + 80 g·L−1催化剂)的工况慢,尤其在反应的初始阶段。该工况的脱色反应也符合拟一级动力学方程,拟合度较高。根据XPS测试结果,相比新制备的催化剂,用过720 min的催化剂表面含有较高比例的化学吸附氧(表5)。在反应中加入催化剂时,催化剂表面的氧空位含量增加,化学吸附氧含量增加,反应中Ni(II)和Ni(III)的可变化合价使催化剂具有很好的电子转移特性,这有利于物理吸附氧捕获电子形成化学吸附氧(式(7))。化学吸附氧和物理吸附氧分子能转化为具有强氧化性的原子氧(式(8)和式(9)),从而使催化剂具有较高的活性。
-
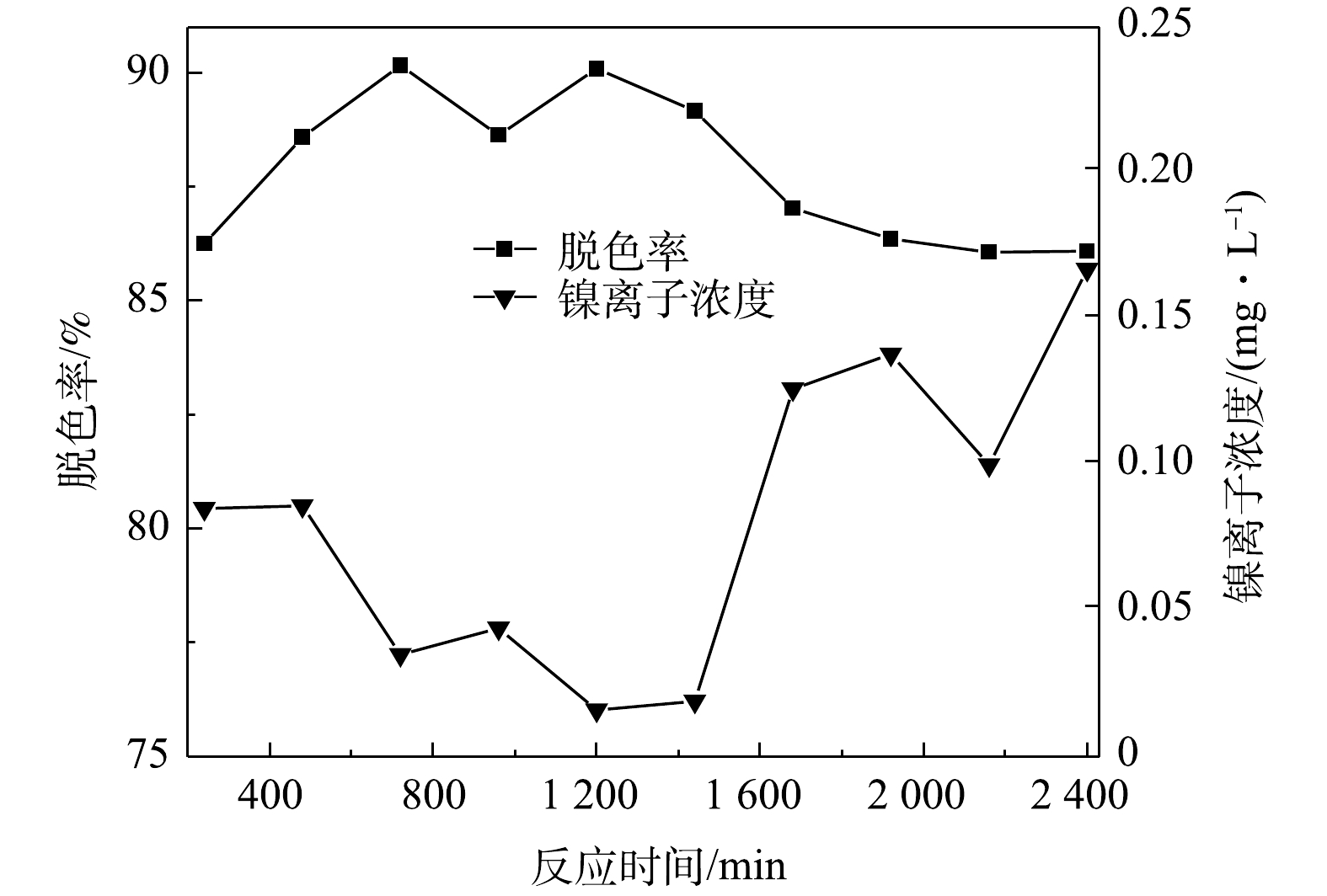
测定溶液在连续流反应器运行不同时间下的脱色率以及处理后出水中镍离子浓度,结果如图9所示。由图9可以看出,当初始pH为9、停留时间为120 min、初始有效氯浓度为120 mg·L−1、初始染料浓度为200 mg·L−1、催化剂投加量为400 g·L−1时,活性艳红K-2BP的脱色率稳定在85%。但出水中的镍离子浓度随着运行时间的增长而逐渐升高,从1 680 min开始,反应器出水中的镍离子浓度就超出了污水排放标准(DB31/199−2018)的限值(0.1 mg·L−1)。随着染料的降解,溶液的pH有所下降,催化剂表面的氢氧化镍与溶液中的H+反应生成的Ni2+,从而从催化剂表面溶出。此外,因为染料的降解导致混合溶液的氧化性有所降低,Ni(Ⅲ)/Ni(Ⅱ)值略微降低,不利于NiOOH镍形态的固定和抑制镍离子的溶出。此现象由下文(2.6节)XPS的分析结果也得到了证实。为了降低出水中镍离子的浓度,决定采用缩短反应器内停留时间、提高初始有效氯浓度和初始pH优化连续流运行条件。
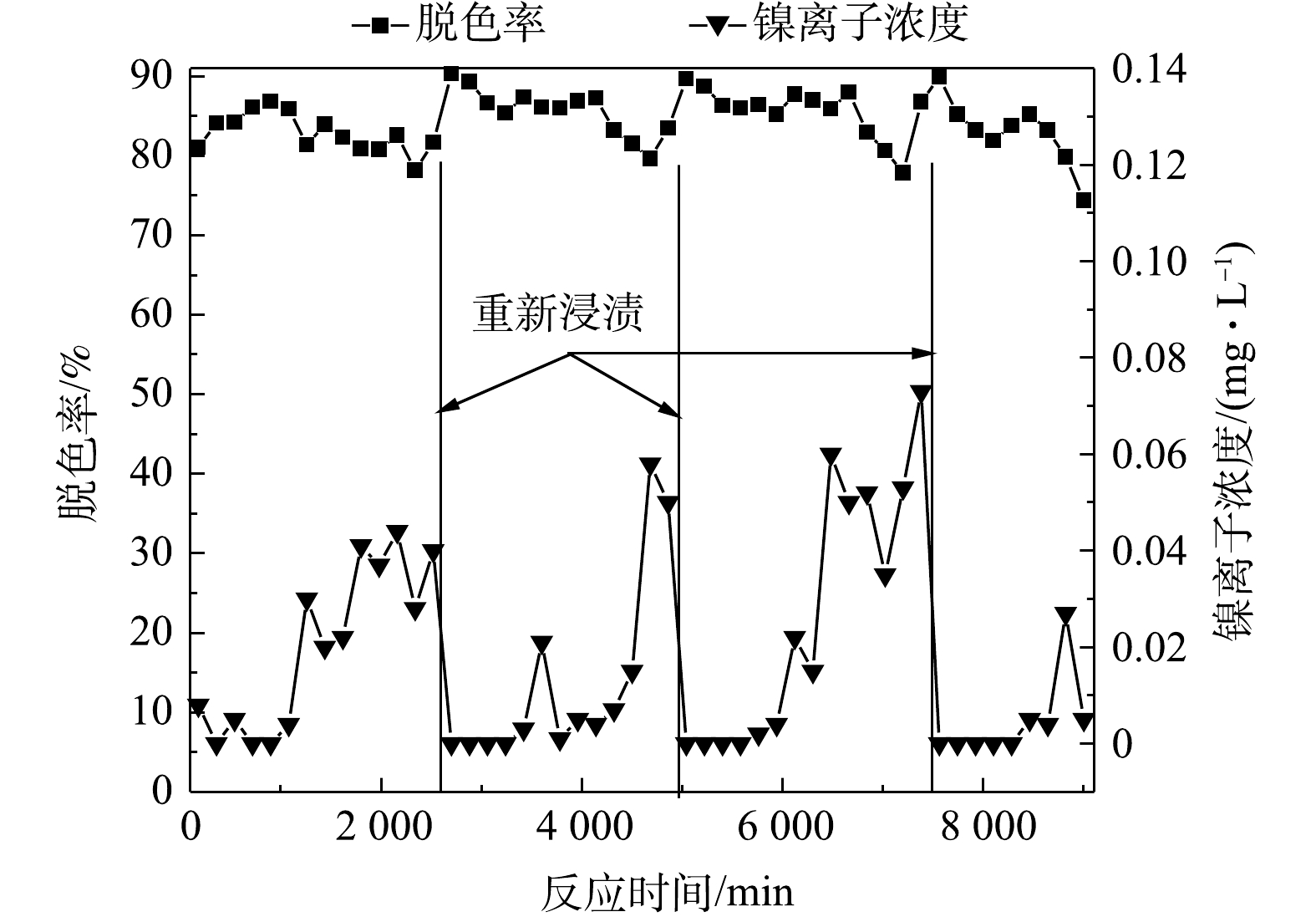
由图10可看出,把反应器内的停留时间缩短到60 min、溶液中初始有效氯浓度提升到200 mg·L−1、初始pH提升到11时,连续流反应器出水的脱色率仍然保持在80%以上,且固镍情况得到明显改善。反应初始时,出水中镍离子浓度基本保持在0.01 mg·L−1以下,远低于污水排放的限值;在运行至1 800 min时,镍离子浓度迅速增大且稳定在约0.04 mg·L−1;在运行至2 520 min时,把催化剂小球从反应器里捞出,用蒸馏水冲洗、烘干,采用次氯酸钠稀溶液浸渍再生催化剂,把恢复活性的催化剂放回反应器中重新开始运行,出水中镍离子浓度降到0 mg·L−1,然后再随着实验的进行慢慢升高;当运行至4 860 min时,镍离子浓度迅速增大且稳定在0.05 mg·L−1,再次采用次氯酸钠稀溶液浸渍催化剂,浸渍后反应器出水中镍离子浓度降低到0 mg·L−1,随后又慢慢增加;当反应至7 380 min后,镍离子浓度迅速升高且稳定在0.07 mg·L−1,再次采用次氯酸钠稀溶液浸渍催化剂。针对该工艺出水,如果水质要求高的话,可以考虑采用化学沉淀法强化混凝工艺去除处理后水样中的微量镍离子[21]。连续流实验出水色度低于30倍,COD为40.7 mg·L−1,达到纺织染整工业水污染物排放标准(GB 4287-2012)。次氯酸钠稀溶液浸渍的频率为6~7 d·次−1。2次再生后的催化剂放在反应器内运行数天,出水中漂浮着少量从催化剂表面脱落的黑色微颗粒,少量微颗粒的存在不影响印染废水的脱色反应,可以采用100目孔径的尼龙滤布去除这些颗粒。把3次浸渍再生后的催化剂放在反应器内运行1 620 min后,印染废水的脱色率降低到74.37%,这是催化层轻微脱落导致的结果,活性氧的产量不能满足染料脱色的需求,此时可以通过次氯酸钠稀溶液浸渍再生催化剂或者采用浸渍法重新制备催化剂。
连续流出水含有较高浓度的有效氯(50 mg·L−1),若直接排放的话,需要采用活性炭过滤、NaHSO3还原等常规方法去除其中的余氯,这样会大大增加水处理的成本。本课题组正开始相关研究工作,以用于减小该工艺出水中余氯的浓度。首先把连续流反应器出水按照一定的比例稀释进水,这样既可以降低印染废水的浓度和色度,也可以把出水中的有效氯利用起来,从而降低次氯酸钠的投加量,降低水处理的运行成本。在不调节pH的情况下,活性艳红/次氯酸钠混合液的pH为10.5~11。随着反应的进行,溶液的pH降低到偏中性,可以直接排放。相比较芬顿法,新工艺具有不需要反复调节pH的优点。
-
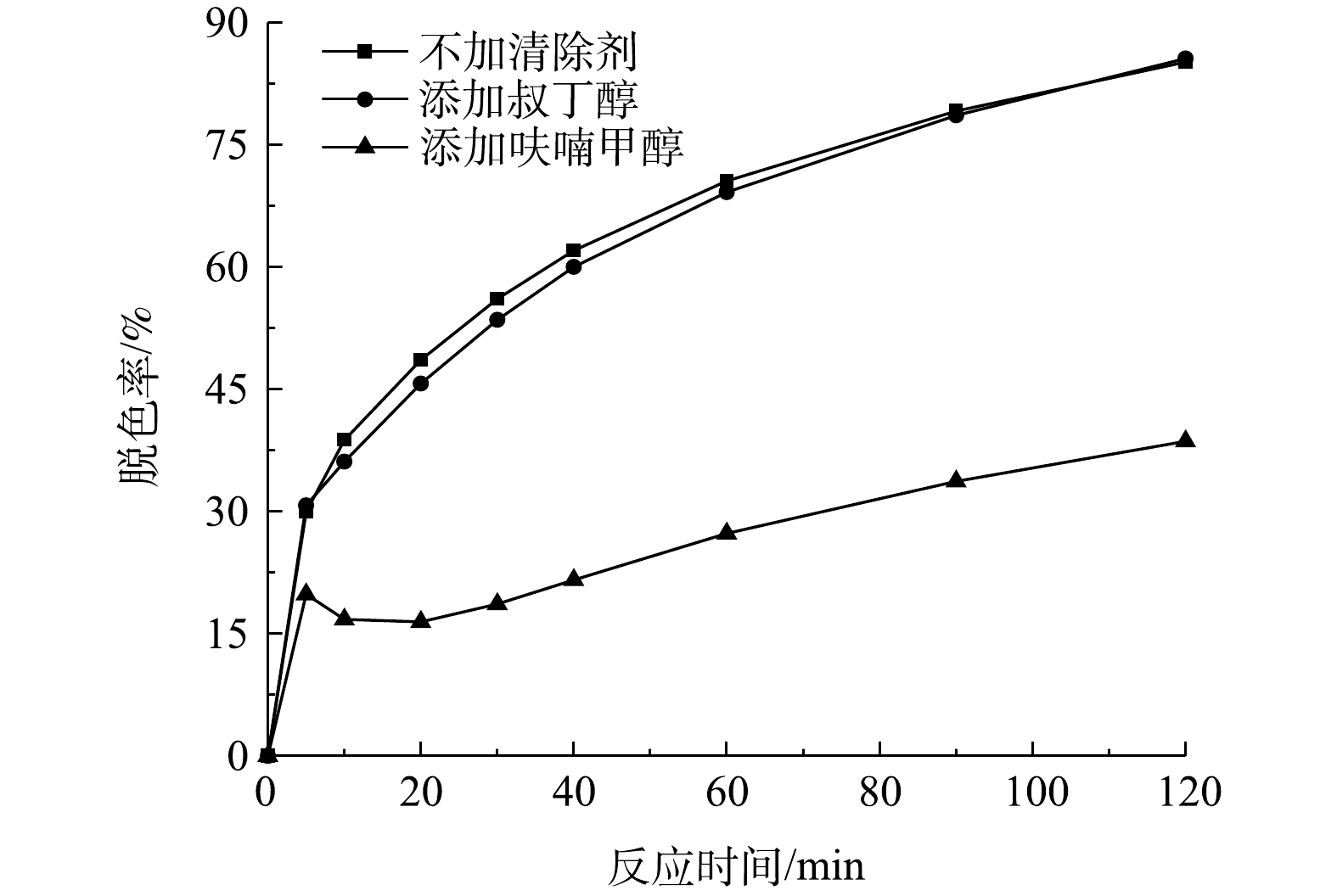
活性氧包括羟基自由基、原子氧、超氧阴离子以及过氧化氢等,其在芬顿法、光催化法、催化臭氧氧化法等高级氧化法降解污染物的过程中起着重要的作用。叔丁醇是一种良好的羟基自由基和硫酸根自由基清除剂,呋喃甲醇能快速地捕获溶液中的原子氧,常被用作原子氧的指示剂[22-23]。在2个浓度为200 mg·L−1和pH为9的模拟印染废水水样中分别添加20 mmol·L−1叔丁醇和呋喃甲醇,再各加入400 g·L−1的催化剂和105 mg·L−1的有效氯,然后观察次氯酸钠催化降解活性艳红K-2BP反应速率的变化情况,并以此来判断在反应中起主导作用的自由基。
由图11可见,加入叔丁醇对活性艳红K-2BP的降解几乎没有影响,而呋喃甲醇对活性艳红K-2BP的脱色效应具有明显的抑制作用,由此可以断定,原子氧在染料降解过程中起着主导作用,羟基自由基和硫酸根自由基在脱色反应中起的作用微乎其微。原子氧是氧的一种高能量形式,其对电子密度大的有机化合物的反应能力要强于基态氧O2和三线态氧O3,比羟基自由基具有更高的选择性,且其自身不能与所有污染物发生反应,但由于具有亲电子性,能在烯型反应中通过夺氢和加成,与不饱和碳碳键迅速发生反应[24]。原子氧能迅速与活性艳红K-2BP中的N=N键发生加成反应,并使之断裂产生NH2,其也能攻击与亚氨基或均三嗪基相连的C—N键,发生夺氢反应进而生成N=C=O[19]。
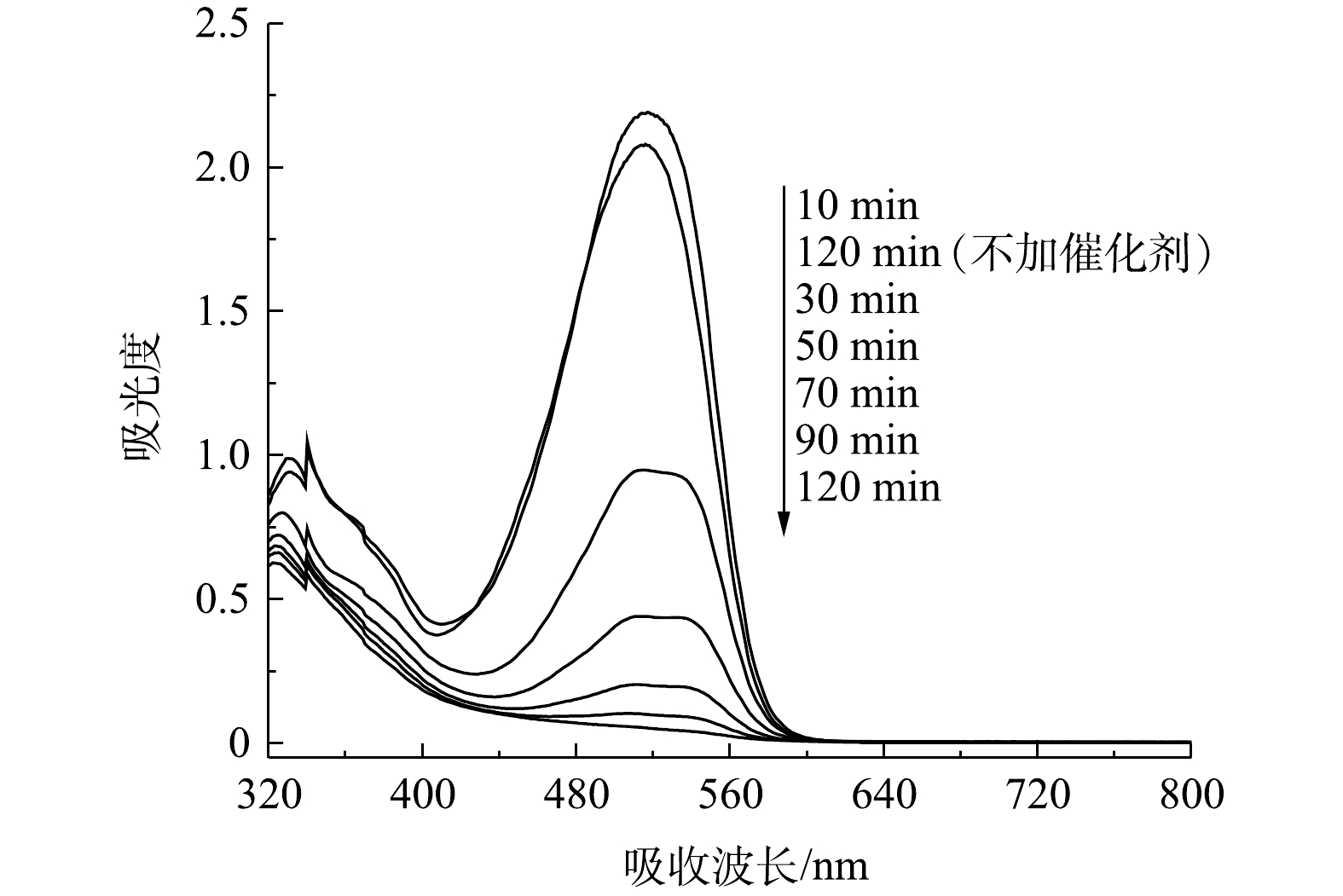
图12是活性艳红K-2BP在不同反应时间和反应条件下的可见扫描光谱图。随着反应的进行,活性艳红K-2BP在534 nm处的偶氮基的最大吸收峰强度迅速降低且发生蓝移,反应120 min时,该峰已基本消失。这说明偶氮染料分子的共轭发色体系被原子氧完全破坏。萘环结构的可见特征吸收峰一般在310 nm处,但其位置会受到附近其他基团的影响发生偏移[25]。322~330 nm处为萘环的吸收峰,峰面积随着反应的进行而减小,但反应至120 min时该特征吸收峰仍然存在,这说明溶液中的萘环没有完全被打开。
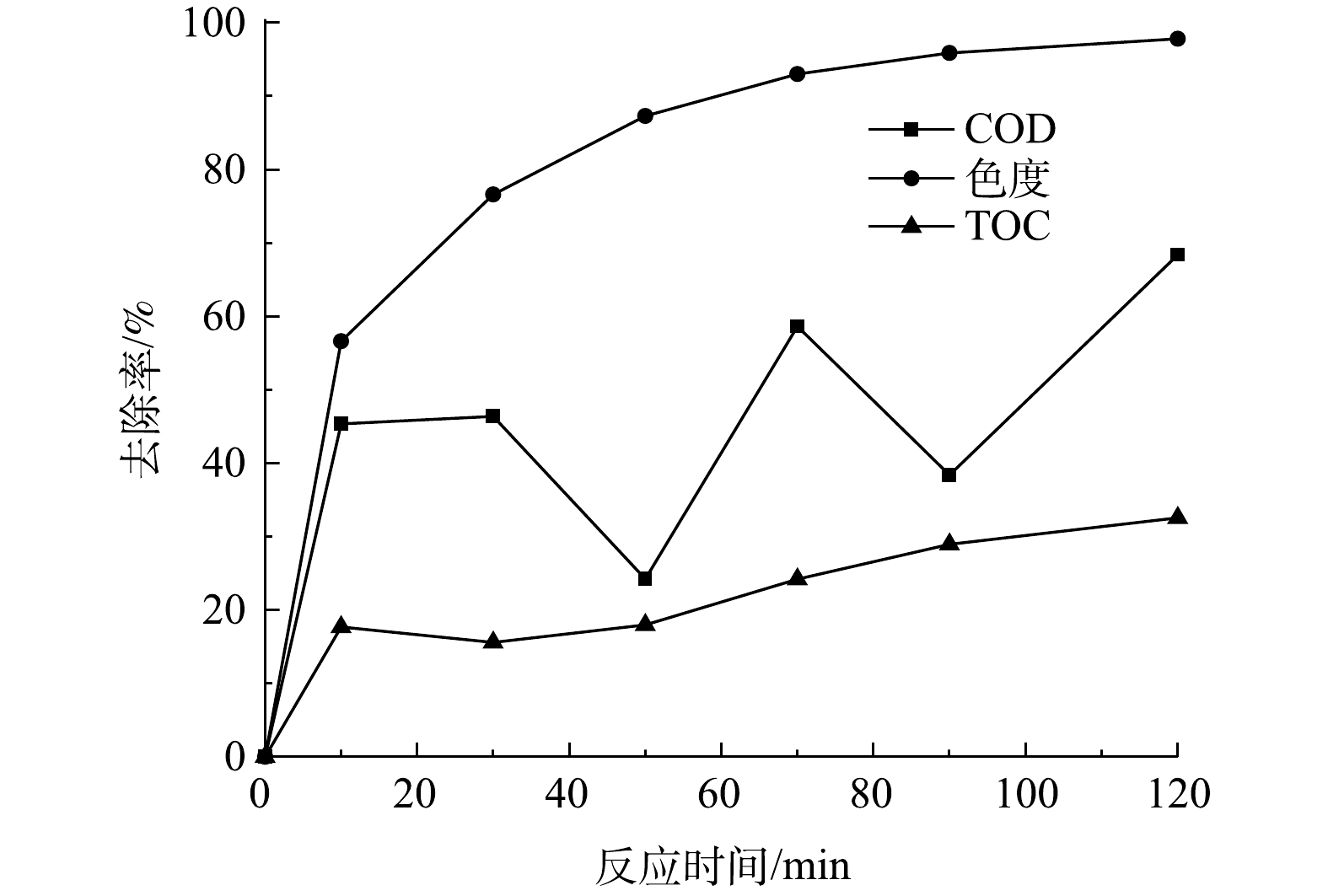
当有效氯浓度为120 mg·L−1、催化剂的投加量为400 g·L−1时,测定COD为67.7 mg·L−1、pH为9、初始浓度为200 mg·L−1活性艳红K-2BP模拟废水在不同反应时间下COD、色度和TOC的去除率,结果如图13所示。由图13可见,活性艳红K-2BP溶液的脱色率大于COD的去除率。在120 min时活性艳红K-2BP脱色率为97.8%,COD去除率为70%,TOC去除率为33%,有机物的矿化程度较低。活性艳红K-2BP被迅速吸附在催化剂表面,COD去除率迅速升高(0~10 min);吸附在催化剂表面的染料分子被原子氧氧化,偶氮键断裂,难降解的染料分子被分解成芳香胺等化合物。较之染料分子本身,芳香胺的COD较高,故溶液的COD值升高,其去除率降低。芳香胺等被进一步氧化分解成小分子的有机物,COD值降低,其去除率增加。染料被慢慢降解,但没有被完全矿化,这应该是氧化剂剂量不够和萘环太难降解而导致的。
处理染料溶液时,本工艺和芬顿法均具有色度去除率高于COD或者TOC的去除率的特点,说明在这2种工艺的作用下,首先破坏染料分子中较弱的发色基团,如Ar−N=N−Ar等(Ar代表芳环),然后再破坏分子中的苯环、萘环等其他键能较高的部位[26];但芬顿法是通过催化分解产生羟基自由基进攻有机物分子,从而使其降解。水解酸化/AO组合工艺能去除活性艳红X-3B印染废水中71%的色度和92.2%的COD,这是因为印染废水脱色主要发生在厌氧阶段,而COD主要通过好氧过程去除[12]。厌氧/好氧工艺处理印染废水时,好氧过程可有效去除厌氧过程中难降解的有机物、厌氧过程中生成的挥发酸(VFAs)和染料代谢产物芳香胺等物质[27]。
采用XPS分析法检测催化剂表面的元素及其相对含量。选取新制备的催化剂、在批量实验中使用过720 min的催化剂和在连续流实验中运行过3 000 min的催化剂作为检测样品。由表5可以看出,催化剂表面氧元素的含量最大,3个样品中氧元素的含量都在80%以上。较之新制备的催化剂,做过连续流实验的催化剂样品含氧量略低,这主要是因为氧被消耗在降解活性艳红K-2BP上,使其溶液脱色。531.46 eV左右的峰为化学吸附氧引起的[28]。化学吸附氧较为活跃,在氧化过程中发挥了巨大作用。YANG等[29]发现,化学吸附氧含量的提升有利于改善催化剂的性能。855.79~856.14 eV的峰是由Ni(Ⅱ)引起的,861.59~861.95 eV的峰是由Ni(Ⅲ)引起的[30]。新制的催化剂样品的Ni(Ⅲ)/Ni(Ⅱ)值为0.62,在连续流实验中运行过3 000 min的催化剂样品的比值为0.46,Ni(Ⅲ)的相对含量减少,Ni(Ⅱ)的含量增加,也就是说,催化剂的催化性能略有降低。与批量实验用过720 min的催化剂比较,新制备的催化剂表面化学吸附氧占比略低。图10所示连续流实验结果表明,催化剂可以通过在NaClO或者过硫酸钠稀溶液里浸渍再生,迅速恢复其催化性能。
图14为催化剂的SEM照片。由图14可见,催化剂的表面比较平整,镍的沉淀物较为均匀且致密地包裹在氧化铝小球表面。因此,镍离子不容易渗透到氧化铝小球里面,制得的复合催化剂在使用过程中也基本不会导致镍离子的溶出,造成二次污染。
2.1. 染料浓度对活性艳红K-2BP溶液脱色率的影响
2.2. 有效氯浓度对活性艳红K-2BP溶液脱色率的影响
2.3. pH对活性艳红K-2BP溶液脱色率的的影响
2.4. 催化剂投加量的影响
2.5. 连续流实验
2.6. 机理研究
-
1)次氯酸钠催化氧化对活性艳红K-2BP废水具有显著的脱色效果,K-2BP的脱色降解过程符合拟一级动力学方程,反应速率常数随着初始染料浓度的增加而降低,随着有效氯浓度和pH的增加而升高。
2)在连续流实验中,活性艳红K-2BP的脱色率稳定在80%以上。当初始pH为11时,催化剂固镍效果好,NaClO和再生的催化剂组成的体系也对K-2BP具有显著的脱色效果,出水中镍的溶出量较低。
3)自由基清除剂实验结果表明,原子氧在染料脱色降解实验中起了重要的作用。
4)新制备的催化剂表面形成一层较为均匀的NiOx(OH)y膜,催化层表面的化学吸附氧随着催化剂使用时间的延长先增加后降低。使用过3 000 min的催化剂的催化性能略微降低。
