

 下载:
下载:


-
铬污染土壤因对人体的巨大危害[1-2]而被广泛关注,一直以来许多专家学者都致力于铬污染土壤的还原修复[3-5]。近年来,由于修复后的铬污染土壤出现严重的“返黄”现象,使得修复后土壤的长期稳定性成为限制铬污染土壤修复工作开展的亟待解决的难点问题[6]。
铬污染土壤修复治理通常采用化学还原技术,通过投加化学还原药剂将土壤中的Cr(Ⅵ)还原为Cr(Ⅲ),但经还原后的铬离子仍然留在土壤中,其价态及生物有效性也可能随着环境的变化而改变[7]。部分研究认为,修复后的铬污染土壤“返黄”,主要是由于外部环境改变导致土壤中的Cr(Ⅲ)被重新氧化成Cr(Ⅵ)。APTE等[6]的研究表明,当Cr(OH)3和MnO2共同悬浮在溶液中时,60 d内Cr(Ⅵ)转化率达1%,且有氧条件下更高。祁光霞等[8]研究发现,增加土壤中的Mn含量,在一定的养护时间内,土壤中Cr(Ⅵ)含量可增加8.5%~21.6%。以上研究表明,在一定含量氧化剂存在的情况下,土壤中的Cr(Ⅲ)有被氧化为Cr(Ⅵ)的风险,但在正常自然条件下,经还原后的Cr(Ⅲ)能否再被自然氧化物质氧化为Cr(Ⅵ),还需要进一步探究,而目前长期对经还原后的实际Cr(Ⅵ)污染土壤跟踪采样分析的研究还鲜见报道。
本研究选取具有高还原性能的CaS4、土壤修复中广泛使用的FeSO4·7H2O和环境友好的葡萄糖3种化学还原剂[9]对3种不同类型的污染土壤进行还原修复,通过对修复后土壤中Cr(Ⅵ)浓度进行定时取样检测,以评估不同药剂还原不同类型的实际Cr(Ⅵ)污染土壤的长期稳定性。同时设置干燥和淹水2个条件,研究了水分条件对土壤长期稳定性的影响,为Cr(Ⅵ)污染土壤的实际修复提供参考。
全文HTML
-
Cr(Ⅵ)污染土壤主要分为3大类,包括表层含铬渣的渣土混合物、渣土层下的重度污染土壤和重度污染层下面的轻度污染土壤。因此,本研究选取以下3类土壤进行研究:A土取自河南某铬盐厂铬渣堆场,主要为含铬渣的渣土混合物;B土取自内蒙古某铬盐厂铬渣堆场,主要为Cr(Ⅵ)重度污染土壤;C土取自甘肃某铬盐厂铬渣堆场,主要为Cr(Ⅵ)轻度污染土壤。样品采集后风干、过2 mm孔径的筛网备用。
实验试剂:FeSO4·7H2O、CaS4、葡萄糖均为分析纯。
-
分别添加一定比例的CaS4、FeSO4·7H2O、葡萄糖对3种土壤进行还原,还原后的土壤均装在50 mL塑料小瓶中,土壤放置厚度为2.5 cm,开盖放置于室内,定时取样检测。各药剂还原投加比例如下:A土,CaS4(理论投加量的2倍)、FeSO4·7H2O(理论投加量的6倍)、葡萄糖(与土的质量比为0.4∶1);B土,CaS4 (理论投加量的1.5倍)、FeSO4·7H2O(理论投加量的2倍)、葡萄糖(与土的质量比为0.2∶1);C土,CaS4 (理论投加量的2倍)、FeSO4·7H2O(理论投加量的3倍)、葡萄糖(与土的质量比为0.02∶1)。以上比例针对3种土壤具有较好的还原效率和经济效益,由还原处理实验[10]得出。实验设置2个平行,结果取平均值。
称取土壤样品90 g放入1 L锥形瓶中,加入900 mL还原药剂与水的混合液,翻转振荡器振荡8 h后,固液分离。所有分离后的土壤晾干混匀备用。
将还原后的土壤均装在50 mL塑料小瓶中,加水淹没土壤,盖上盖子密闭,放于室内待测。其余实验步骤同上。
-
应用马尔文2000激光粒度仪测定土样粒径,X射线衍射(XRD)等来判定3种Cr(Ⅵ)污染土壤的物相组成。XRD表征条件:CuKa 靶,电压为40 kV,电流为40 mA,扫描速度为2(°)·min−1,光源波长为1.54 nm,角度为5°~90°。酸溶态Cr(Ⅵ)参照《固体废物浸出毒性浸出方法硫酸硝酸法》 (HJ/T 299-2007)[11],水溶态Cr(Ⅵ)浸出参照《固体废物浸出毒性浸出方法翻转法》(GB 5086.1-1997)[12],水溶液中Cr(Ⅵ)的测定采用《水质 六价铬的测定 二苯碳酰二肼分光光度法》(GB 7467-1987)[13],土壤中Cr(Ⅵ)的测定参照《碱消解/火焰原子吸收分光光度法》(HJ 687-2014)[14]。
1.1. 实验原料
1.2. 实验方法
1.3. 分析方法
-
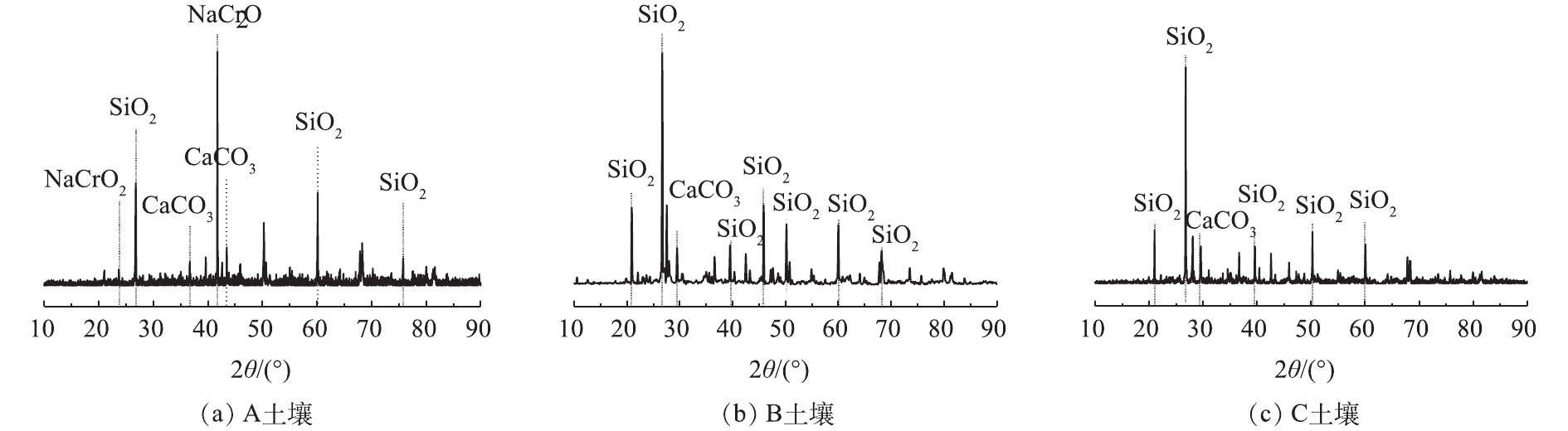
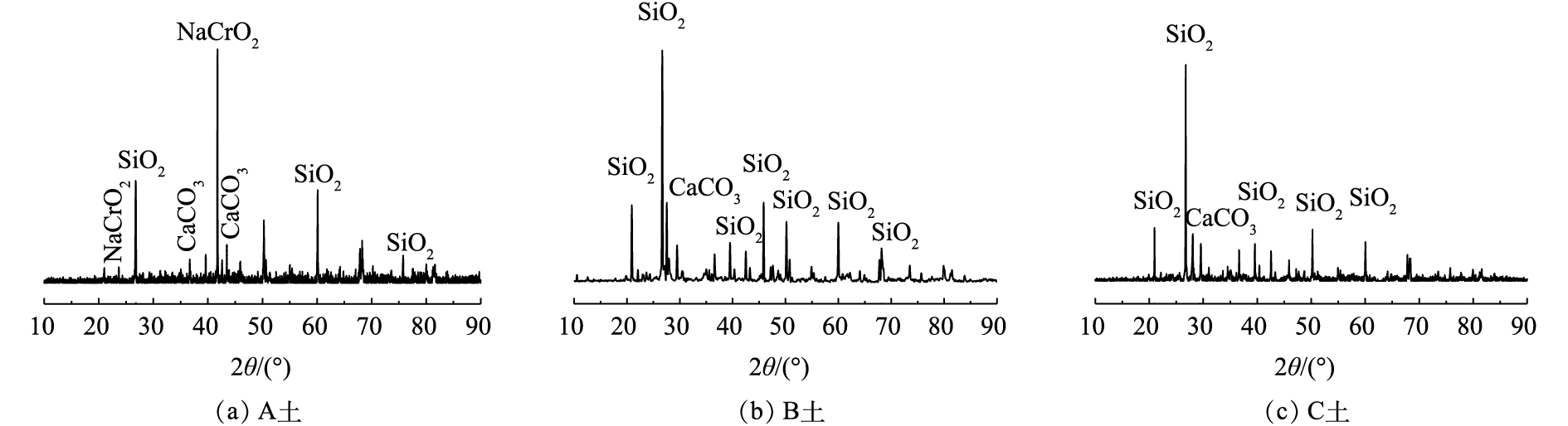
对供试土壤进行X射线衍射分析,分析结果如图1所示。可以看出,A土为混有铬渣的土壤,其XRD图谱中最强的特征峰在41.74°(JCPDS PDF#88-1676),这表明存在Cr(Ⅲ)化合物NaCrO2,这是铬渣混土的一个典型特征;B土和C土是Cr(Ⅵ)污染土壤,二者XRD图谱较为相似,并没有出现铬的特征峰,说明Cr(Ⅵ)污染土壤中以晶态形式存在的铬较少。
3种供试土壤的粒径分布等物理特性如表1所示。A土中粉粒和砂粒基本各占一半,B土以粉粒和砂粒为主,且砂粒占比更高,C土则是粉粒占到了绝大多数;三者相比,C土相对粒径较小,A土居中,B土粒径较大,A土和B土的质地为砂壤土,C土为粉壤土。表2为供试土壤中铬的含量和形态测试结果,水溶态Cr(Ⅵ)和酸溶态Cr(Ⅵ)是土壤中Cr(Ⅵ)在不同外部浸出环境下的可溶态形式,由表2可以看出,B土和C土中Cr(Ⅵ)绝大部分以可溶态形式存在,这是Cr(Ⅵ)污染土壤直接采取淋洗、还原稳定化修复的重要原因[15];而A土中水溶态Cr(Ⅵ)和酸溶态Cr(Ⅵ)占比均不到50%,说明A土中还有一半左右的Cr(Ⅵ)较难溶出,这也是渣土混合物修复难度大的原因[16]。
-
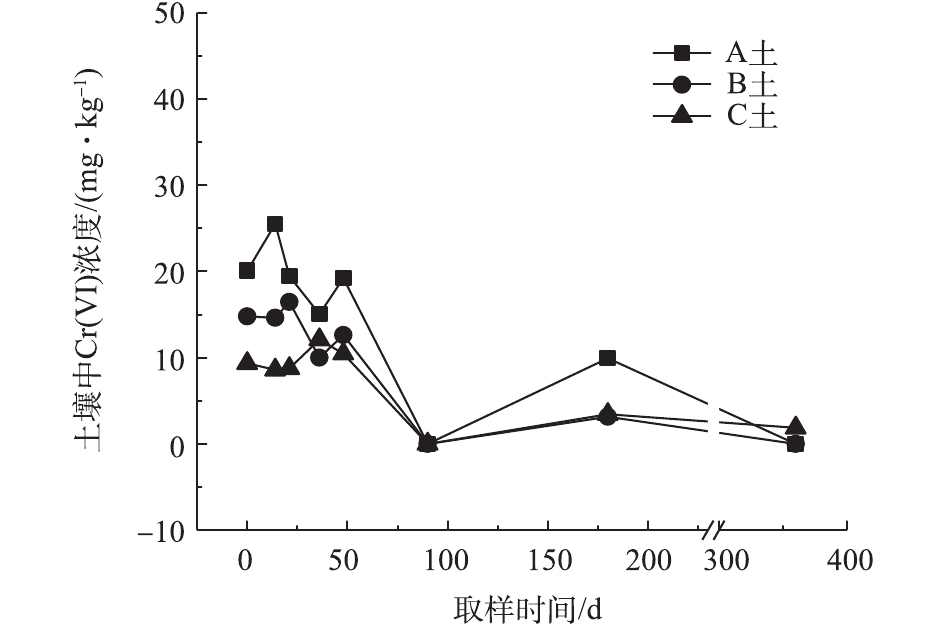
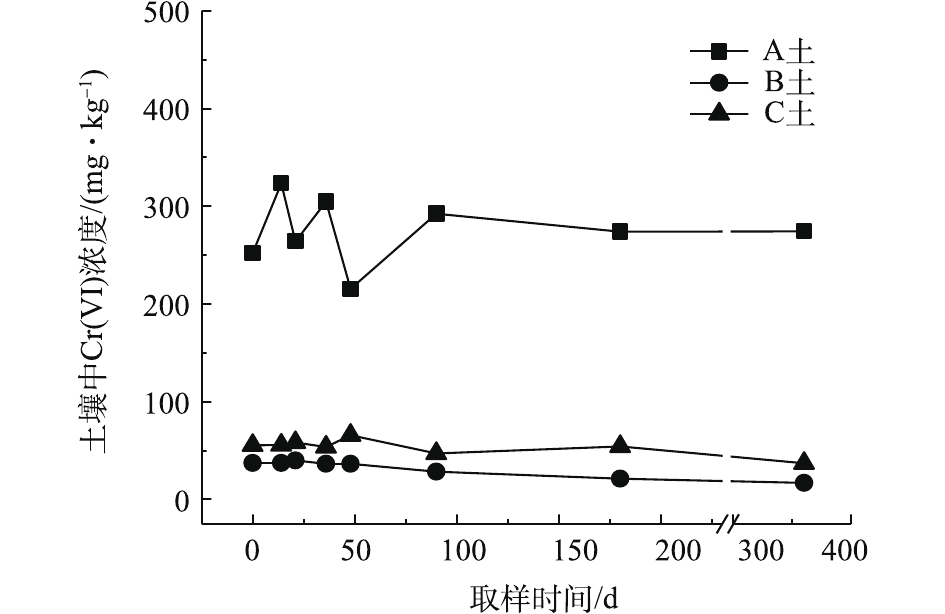
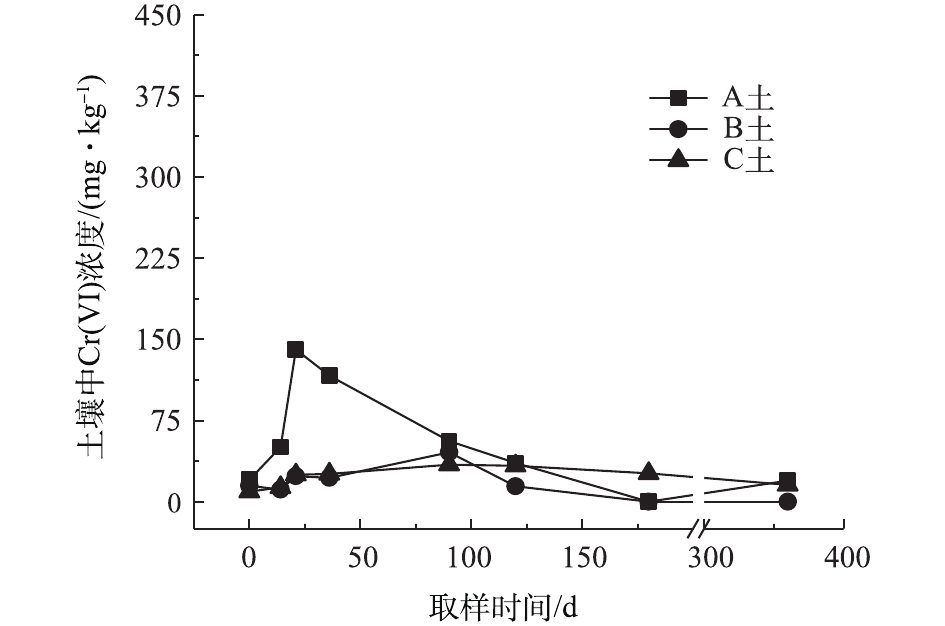
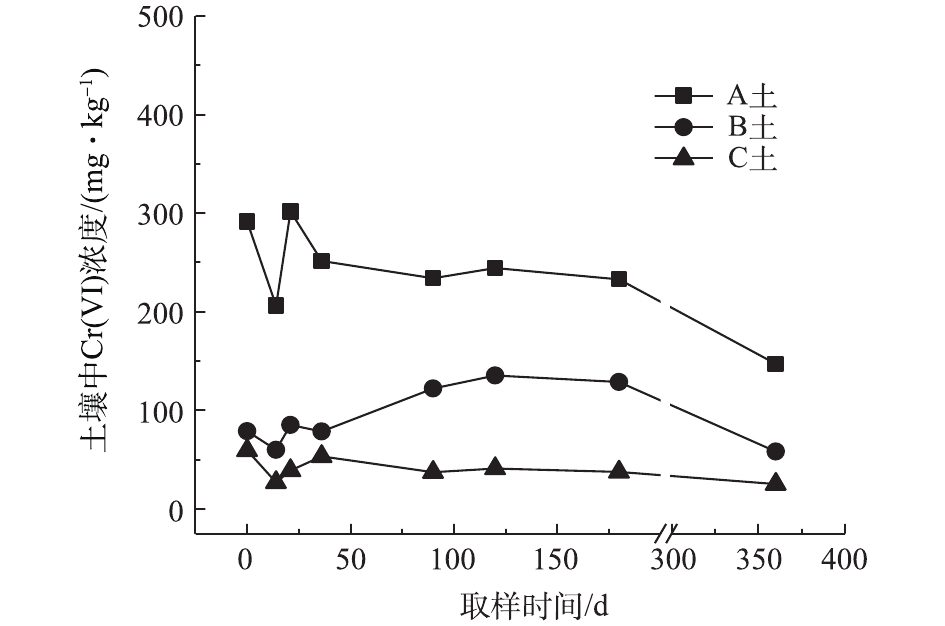
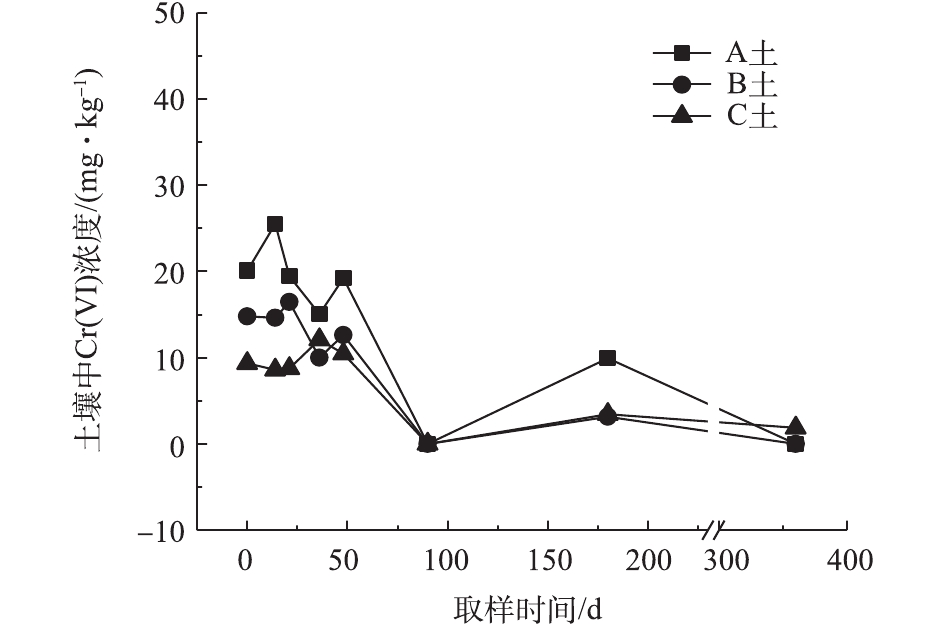
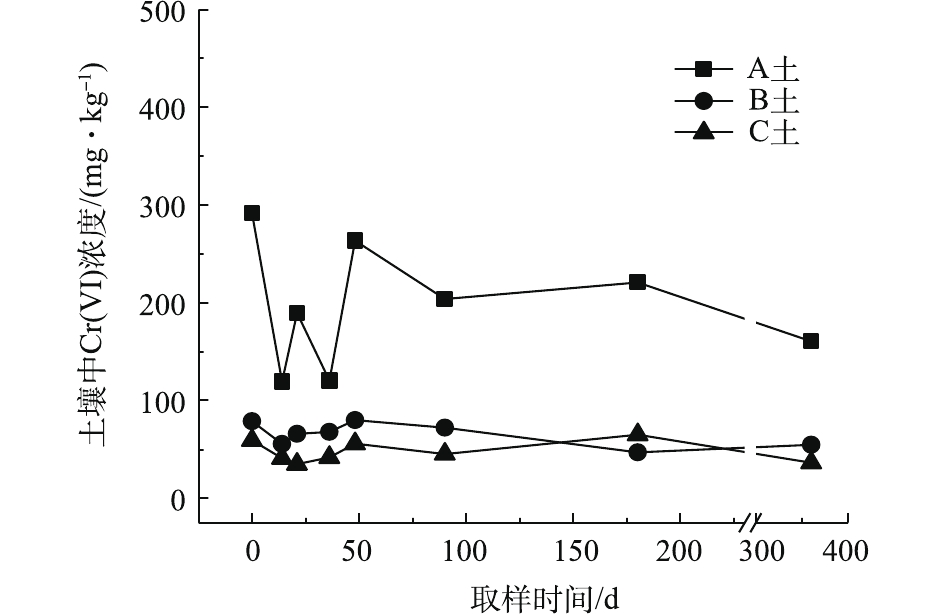
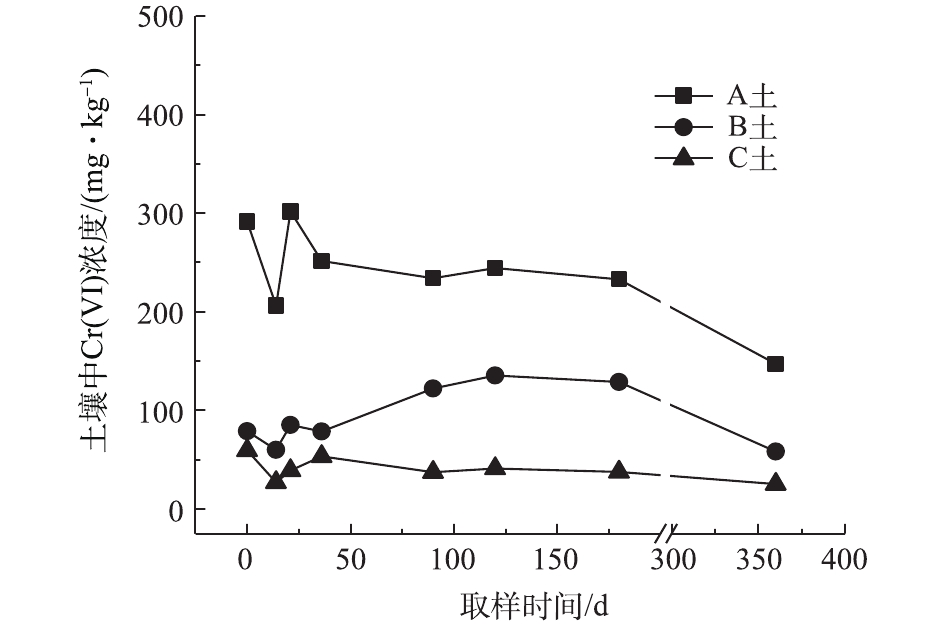
对不同药剂还原后的Cr(Ⅵ)污染土壤定时取样检测,土壤中Cr(Ⅵ)浓度变化情况如图2~图4所示。由图2~图4可知,投加CaS4药剂处理后的3种土壤中Cr(Ⅵ)含量接近且均较低,说明3种土壤中Cr(Ⅵ)经CaS4还原比较彻底且反应程度较为一致;在360 d的跟踪采样实验周期内,投加CaS4还原处理的3种土壤中Cr(Ⅵ)浓度均呈较稳定的状态,其浓度波动幅度均低于20 mg·kg−1。投加FeSO4·7H2O处理后的3种土壤中,残留的Cr(Ⅵ)浓度比CaS4处理结果要高,特别是处理后的A土中Cr(Ⅵ)浓度比B土和C高,说明FeSO4·7H2O的还原效果较CaS4差,尤其是对渣土混合物的还原效果更不理想。在跟踪实验周期内,B土和C土Cr(Ⅵ)浓度较为稳定,其波动幅度分别约为35 mg·kg−1和30 mg·kg−1,但A土Cr(Ⅵ)分析结果波动幅度较大,约为170 mg·kg−1。一方面,由图1知A土铬渣中含有晶格态Cr(Ⅵ),其赋存形态特殊,不易被彻底还原,在长期堆存过程中逐步释放溶出,对采样分析结果产生一定影响;另一方面,由于A土中混有的高Cr(Ⅵ)含量铬渣分布不均匀,导致取样均质性差异过大进而影响分析结果。从波动趋势来看,前期波动较后期大,随着取样时间的延长,土壤中Cr(Ⅵ)含量逐渐趋于稳定,土壤中残留少量的还原药剂也逐渐失效,不对土壤中的Cr(Ⅵ)产生影响。投加葡萄糖药剂的实验结果和投加FeSO4·7H2O较为相似,药剂还原Cr(Ⅵ)的程度低于CaS4,且对A土还原效果更低;在实验周期内,B土和C土中Cr(Ⅵ)较为稳定,其波动幅度分别约为20 mg·kg−1和30 mg·kg−1,而A土Cr(Ⅵ)波动幅度约为150 mg·kg−1。
经药剂还原后,土壤中的Cr(Ⅵ)浓度变化结果表明:在干燥条件下,CaS4还原后的Cr(Ⅵ)污染土壤均具有较强的稳定性,针对B土、C土这类以易溶态Cr(Ⅵ)为主的污染土壤,添加FeSO4·7H2O和葡萄糖还原后,长期稳定性相比CaS4较好;针对A土渣土混合物,添加3种还原剂的处理效果稳定性较差,一方面是由于A土所含Cr(Ⅵ)形态差异较大,使得供试样品的均质性和稳定性都比较差,另一方面,随着实验周期的延长,供试环境条件的变化会使得未被还原的晶格态Cr(Ⅵ)向环境中逐步释放,对还原后土壤的长期稳定性产生影响。
-
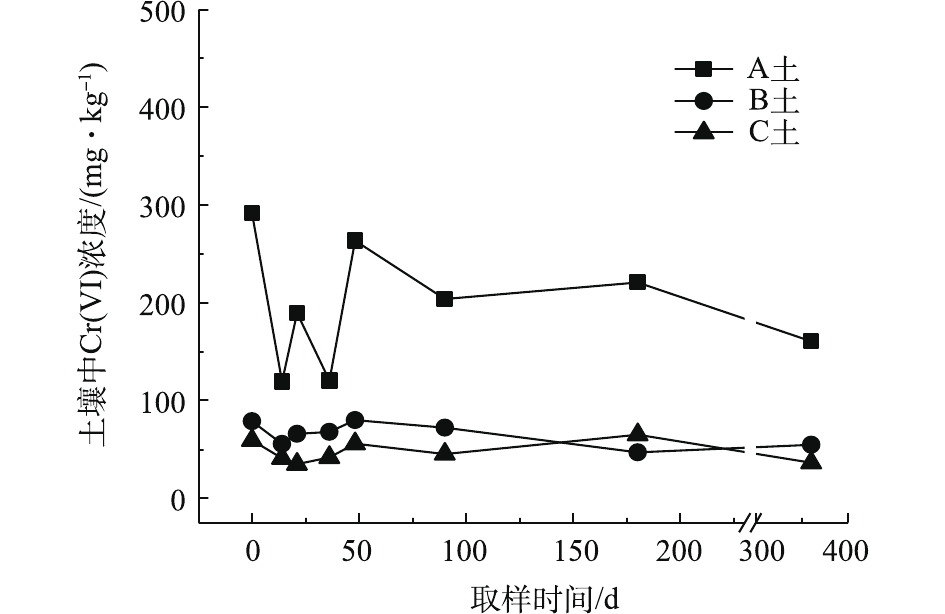
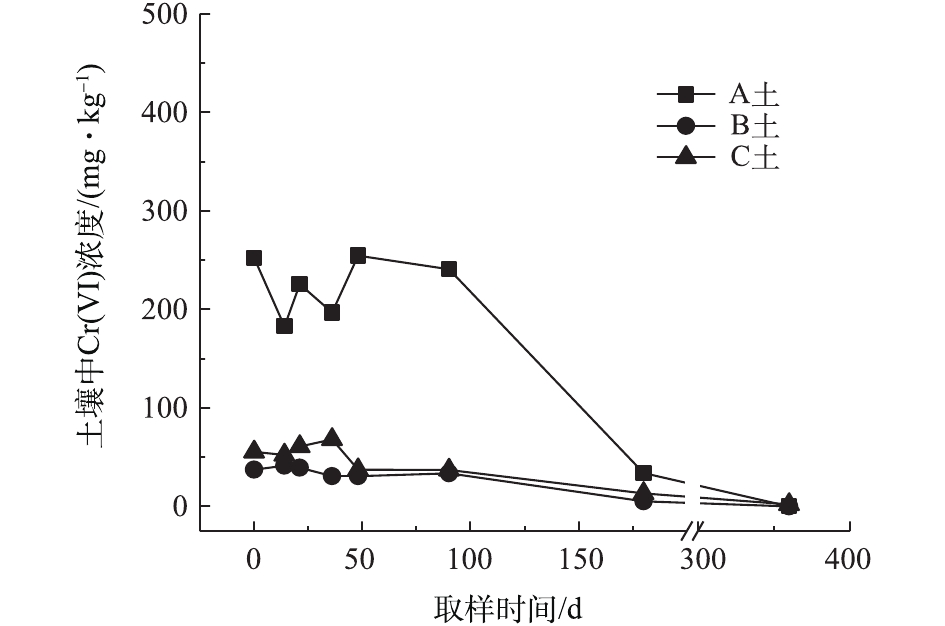
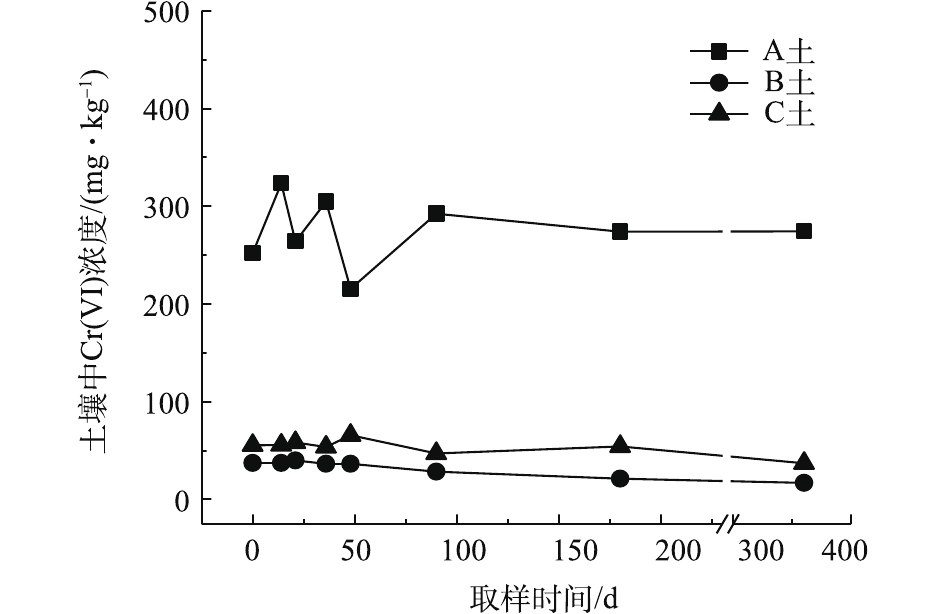
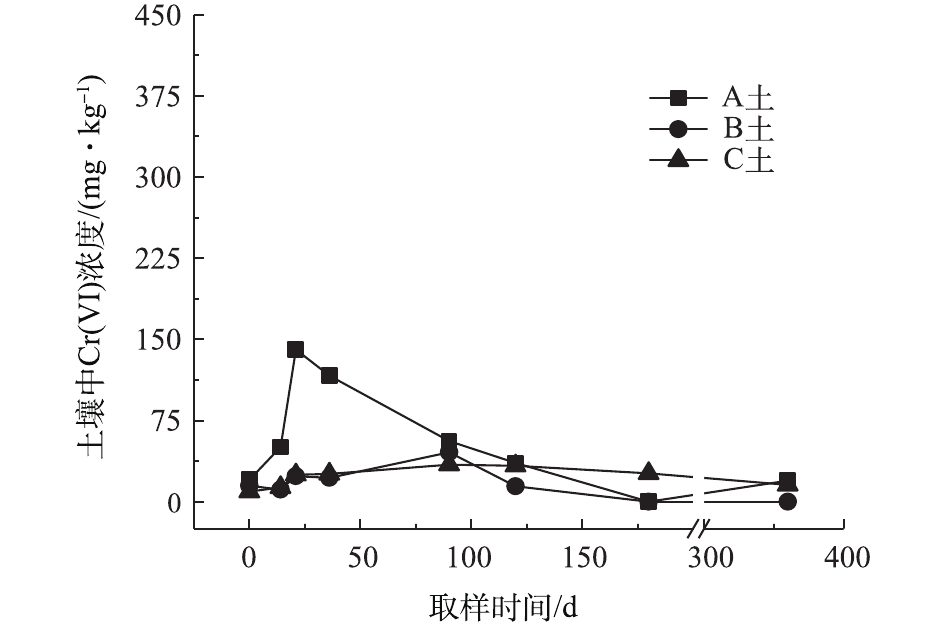
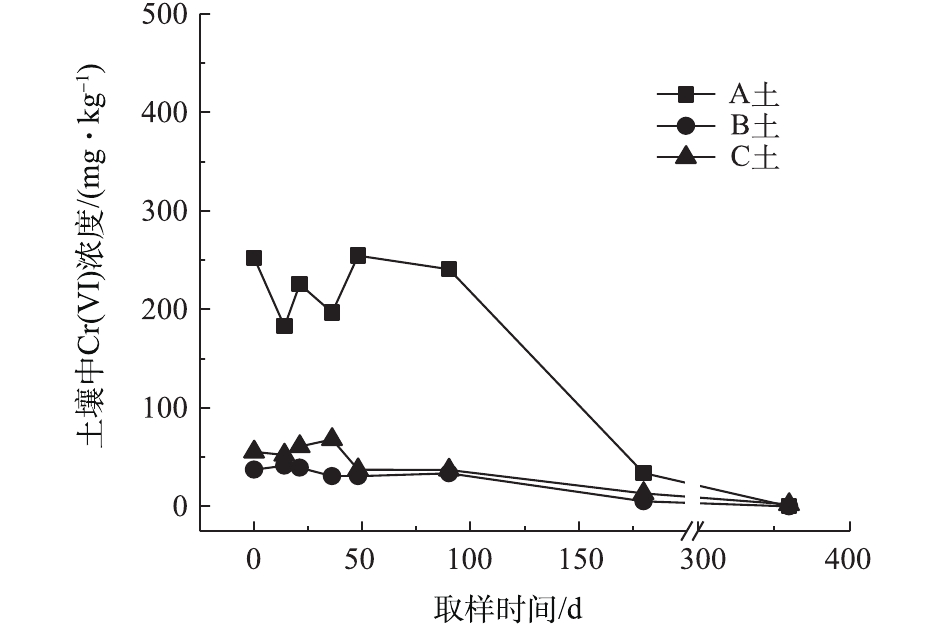
对不同药剂还原后的Cr(Ⅵ)污染土壤加水淹没密闭,定时取样并检测其中的Cr(Ⅵ)浓度,土壤中Cr(Ⅵ)浓度变化情况如图5~图7所示。由图5~图7可知,在淹水条件下,投加CaS4还原后3种土壤Cr(Ⅵ)浓度都能降到50 mg·kg−1以下,A土中由于含有难溶出的晶格态Cr(Ⅵ),在实验周期内,Cr(Ⅵ) 浓度波动幅度达到120 mg·kg−1,但在最终采样时,3种土壤的还原反应程度较为一致;在360 d的跟踪采样实验周期内,投加CaS4还原处理的3种土壤中,Cr(Ⅵ) 浓度经过一段时间波动之后,均呈现一定的下降趋势, 其中,A土中Cr(Ⅵ) 浓度的下降趋势尤为明显。投加FeSO4·7H2O还原后的3种土壤Cr(Ⅵ)浓度总体呈现下降趋势,360 d时的采样分析结果表明,3种土壤的Cr(Ⅵ)浓度相比实验起始阶段均有所降低。这可能是因为在淹水条件下,土壤中Cr(Ⅵ)能持续溶出且被还原;而在密闭条件下,体系处于少氧或厌氧状态,过量的Fe(Ⅱ)不易被氧化成Fe (Ⅲ),实验体系保持还原性能,可将持续溶出的Cr(Ⅵ)还原为Cr(Ⅲ),最终使得土壤中Cr(Ⅵ)浓度持续降低。投加FeSO4·7H2O还原后的A土中由于含有不易溶出的晶格态Cr(Ⅵ)渣土,其Cr(Ⅵ)溶出反应周期更长,前后降低幅度更大,约为150 mg·kg−1。在淹水和密闭条件下,投加葡糖糖还原后的A土、B土、C土中Cr(Ⅵ)浓度从起始阶段的252、37、55 mg·kg−1降至无法被检出的水平。这说明葡萄糖等弱还原性的有机物针对Cr(Ⅵ)有较好的长效性,不仅能促进土壤中难溶出的Cr(Ⅵ)持续溶出并被还原,在较长时间内确保Cr(Ⅵ)污染土壤保持稳定;而且还针对A土渣土中难溶出晶格态Cr(Ⅵ)的溶出和还原有促进作用,使得A土在实验周期内也能得到最大程度的还原处理。
综上所述,淹水密闭条件下,经过3种还原药剂处理的A土、B土、C土,其Cr(Ⅵ)含量随时间呈现一定的下降趋势。特别当葡萄糖作为一种弱还原剂时,在实验条件下,其呈现较好的长效性,针对3种土壤的处理效果均较好。另外,在淹水条件下,A土由于所含铬渣与土壤污染特性的差异,使得其还原稳定性较B土、C土差,实验周期内,其Cr(Ⅵ)浓度波动幅度都相对较大。
-
土壤氧化还原电位(Eh)的高低取决于土壤溶液中氧化态和还原态物质的相对浓度,影响因素主要有土壤水分状况和通气性等。土壤的Eh值可从还原状况的−200~300 mV到氧化状况的700 mV[17],实验过程中,测得干旱条件和淹水条件下3种土壤经3种药剂还原后的pH均低于9.15,氧化还原电位均低于200 mV,根据铬转化的Eh-pH关系图[18]可知,在土壤pH为 6~9的条件下,Cr(Ⅲ)和Cr(Ⅵ)转化的氧化还原电位为350~650 mV,而实验测得土壤的Eh远低于该范围值。因此,在本研究模拟条件下,经还原后土壤的Eh难以将Cr(Ⅲ)氧化成Cr(Ⅵ),土壤中的Cr(Ⅲ)是比较稳定的。
-
从还原试剂角度分析,投加3种还原试剂还原后土壤中Cr(Ⅵ)稳定性的强弱顺序为CaS4>葡萄糖>FeSO4·7H2O;从污染土壤类型分析,铬渣混土因混有铬渣,晶格态Cr(Ⅵ)含量较高,使得其稳定性较水溶态和酸溶态Cr(Ⅵ)占比高的土壤较弱。对淹水和干燥密闭2种条件下土壤中Cr(Ⅵ)稳定性比较可知,淹水密闭条件下,经药剂还原后的土壤稳定性较差。由于Cr(Ⅵ)水溶性极强,淹水环境不仅为Cr(Ⅵ)提供了良好的溶出环境,且为铁、硫等离子提供了反应原料与场所,使得实验过程中发生的反应较干燥条件更为复杂。在淹水条件实验过程中,由于密闭少氧或缺氧,故导致土壤中过量的还原剂保持长期有效,并未被空气所氧化,从而使得土壤中晶格态Cr(Ⅵ)等缓慢溶出并持续被还原,实验周期内土壤中Cr(Ⅵ)含量呈现出下降的趋势。
综上所述,在实际Cr(Ⅵ)污染土壤的修复治理过程中,若采用CaS4、FeSO4·7H2O进行还原,在进行修复后,土壤的养护过程中应尽量降低土壤的含水量,最大限度地减少Cr(Ⅵ)的溶出。葡萄糖在实际的土壤修复应用中还较少,但通过研究发现,淹水密闭条件下葡萄糖能显著降低包括铬渣混土在内的污染土壤中Cr(Ⅵ)浓度,而且葡萄糖对土壤危害较小[19],具有较好的应用前景。渣土混合物较单一污染土壤而言,稳定性更差,在实际的土壤修复过程中,应将渣土混合物分开管理,节约成本,同时也降低了土壤“返黄”的风险。
2.1. 污染土壤性质
2.2. 不同药剂还原后Cr(Ⅵ)污染土壤的稳定性评估
2.3. 淹水条件对还原后Cr(Ⅵ)污染土壤稳定性的影响
2.4. 经还原后的Cr(Ⅵ)污染土壤稳定性分析
2.5. 还原试剂类型、污染土壤性质及是否淹水密闭对还原后Cr(Ⅵ)污染土壤稳定性的影响
-
1)针对轻、重污染土壤和渣土混合物,投加CaS4对土壤中Cr(Ⅵ)的还原效果要优于FeSO4·7H2O和葡萄糖。
2)在360 d的跟踪采样周期内,在干燥条件下,3种土壤经CaS4处理后长期稳定性较强,土壤中Cr(Ⅵ)含量随时间波动幅度较小;轻、重污染土壤经FeSO4·7H2O和葡萄糖药剂处理后,Cr(Ⅵ)浓度保持稳定,但渣土混合物在投加FeSO4·7H2O和葡萄糖处理后,Cr(Ⅵ)波动幅度较大;在淹水密闭条件下,3种土壤在投加3种还原药剂后,Cr(Ⅵ)含量都有随时间下降的趋势,特别是渣土混合物在投加FeSO4·7H2O和葡萄糖药剂处理后,Cr(Ⅵ)含量随时间下降趋势明显;与淹水密闭条件相比,经还原修复后的Cr(Ⅵ)污染土壤在干燥条件下具有更强的长期稳定性。
3) Cr(Ⅵ)污染土壤在投加过量还原药剂处理后,在360 d的采样周期内,在土壤中过量还原剂还没有被环境破坏的前提下,土壤中Cr(Ⅵ)保持相对稳定;特别是在有水参与体系反应的条件下,土壤中Cr(Ⅵ)有继续下降的趋势。在修复后更长的时间内,修复后土壤中过量还原剂可能被破坏,此时Cr(Ⅵ)的稳定性还需要进一步探究 。
