






-
印染废水具有色度高、有机物含量高、成分复杂和可生化性能差等特点,是一种难处理的工业废水[1]。江苏是印染产业大省,近10年来涌现出一批具有区域集聚的印染产业园,2008年印染产能位居全国第二,2011年位居全国第三。全省960多家重点印染企业有60%位于太湖流域,印染废水和COD的排放量分别占全省印染行业的75.3%和72.3%,对太湖流域产生较大污染[2-4]。为控制太湖水体富营养化,维护生态平衡,促进沿湖地区社会经济和环境的协调发展,江苏省出台了《太湖地区城镇污水处理厂及重点工业行业主要污染物排放限值》(DB 32/1072-2018)[5],并即将在全省施行。这一高排放标准将纺织染整行业列为重点工业行业,COD、总氮和总磷的直接排放标准分别为60、12和0.5 mg·L−1,对其排放有了更高的要求。因此,大量的印染企业面临原有水处理设施提标改造的需求。
常用的印染废水处理方法为结合物化及生化的二级处理工艺,可去除废水中的大部分污染物,但出水的色度、COD等指标不能满足新标准的要求[6]。随着研究的深入,各类污染物的强化去除工艺也日益增多。有研究[7-9]表明,生物吸附池可利用细菌的絮凝吸附作用实现对进水中有机物的高效吸附和代谢降解,快速降低进水中的有机物含量,减轻后端工艺负荷。MBBR工艺是通过投加悬浮载体提高反应器中的生物量及生物种类,从而提高反应器的处理效率,强化有机物降解与好氧硝化作用,提升抗冲击负荷能力[10-13]。硫铁自养反硝化滤池兼顾脱氮和除磷功能,在硫磺上的富集脱氮硫杆菌以单质硫为电子供体将硝酸盐氮还原为氮气,铁屑析出的Fe3+与
PO3−4 -P结合,可有效保障印染废水中氮和磷的去除[14-17]。混凝沉淀工艺的研究[18-19]表明可通过投加絮凝剂强化对磷和有机物的去除。胡溪等[20]和李欣珏[21]提出活性焦在印染废水处理中表现出较好的效果,可有效吸附大部分大分子有机物,对色度和异味也有较好的去除,可作为组合工艺出水指标的保障单元。这些工艺对印染废水中的有机物、氮磷和色度的处理上各有其优势,功能上又相互独立。针对印染废水进水有机物浓度高,可生化性差,色度高,成分复杂等特点,本研究从高标准排放的角度出发,研究了生物吸附/MBBR/混凝沉淀/硫铁自养反硝化/活性焦组合工艺对实际印染废水的处理效果,验证了不同HRT和DO对系统污染物去除的影响,以期实现高标准排放。
-
实验进水为太湖流域某印染集中污水处理厂调节池水,主要水质指标如表1所示。生物吸附池和MBBR的接种污泥均来自该厂好氧池活性污泥,MLVSS/MLSS均值为0.53、生物吸附池HRT为0.5 h、SRT为1 d、DO为0.5~1 mg·L−1、MLSS为5 000~6 000 mg·L−1;MBBR池的SRT为25 d、DO为3~5 mg·L−1、MLSS维持在7 000~9 000 mg·L−1;硫铁自养反硝化滤池的填料硫颗粒来自于已挂膜成功的某硫自养反硝化中试反应器,其HRT为4 h;混凝沉淀池中投加25 mg·L−1的PAC;活性焦柱的HRT为2 h。通过改变蠕动泵进水流量和各构筑物标高调整HRT,通过转子流量计改变曝气流量调整DO浓度。
-
实验装置主体采用无色有机玻璃制作,工艺流程如图1所示。生物吸附池为圆柱体状,内径为23 cm,有效容积为6 L。MBBR池为圆柱体状,内径为41 cm,有效容积为50 L。生物吸附池和MBBR池底部设有曝气头进行曝气充氧。混凝沉淀池出水通过蠕动泵注入硫铁自养反硝化滤池,滤池为圆柱体状,内径为10 cm,为使配水均匀及防止堵塞,滤池底部设有10 cm的碎石承托层。填料硫粒径为2~4 mm,孔隙率约为50%。铁屑以塑料球形式包裹,填充比例为20%,有效容积为5 L。活性焦柱为圆柱体状,内径为10 cm,有效容积为5 L,底部设有10 cm的碎石承托层。
-
反应器启动成功后,调整生物吸附池、MBBR池和硫铁自养反硝化滤池的HRT,确定最佳HRT后,在不同DO条件下运行生物吸附池、MBBR池。由于硫铁自养反硝化滤池在运行过程出现除磷不稳定的现象,因此,在32 d时将硫铁自养反硝化滤池调整为纯硫自养反硝化滤池。具体运行条件如表2所示,最终确定生物吸附/MBBR/混凝沉淀/硫铁自养反硝化/活性焦组合工艺的最佳运行特性。
-
COD、
NH+4 -N、NO−3 -N、TN和TP常规指标均采用国家标准方法[22]测定。MLSS和MLVSS采用重量法[23]测定。DO、pH均采用德国WTW手持便携式多参数水质分析仪Multi3430测定。 -
生物吸附/MBBR/混凝沉淀/硫铁自养反硝化/活性焦组合工艺对进水中COD的去除效果如图2所示。由图2(a)可知,进水COD值范围为323~871 mg·L−1,平均值为542 mg·L−1,进水有机负荷变化较大。在装置运行过程中,生物吸附池的出水在40 d左右时相对于其他单元的波动较大,其原因是受到上游企业集体排水影响,污水厂进水水质波动剧烈,生物吸附池污泥受到进水冲击,导致生物吸附池处理效果不稳定,如图2(b)所示。
在装置运行稳定后,调整装置的运行参数。在优化阶段1,将生物吸附池的HRT从0.5 h增加至1 h后,生物吸附池对COD的去除效果得到提升,出水COD值从400 mg·L−1左右降至200 mg·L−1左右。在优化阶段2,继续增加生物吸附池的HRT,出水COD值无明显下降。生物吸附池对污染物的去除分为吸附和降解2个阶段,活性污泥微生物需要与废水在反应器中快速接触30~60 min。当HRT过短时,微生物对污染物的吸附及网捕等作用还未完全,而增加生物吸附池的HRT可给微生物提供更长的吸附时间,从而可有效提升反应器的处理效率[24-27]。在有氧条件下,微生物还可将吸附的有机物分解成小分子物质。因此,提高DO浓度可增强微生物对有机物的降解能力。所以,在优化阶段2调整了生物吸附池的DO。将DO从0.5 mg·L−1增加至1 mg·L−1,出水COD从约200 mg·L−1降至150 mg·L−1左右。在优化阶段3,继续增加生物吸附池的DO,出水COD值无明显下降。因此,选择DO为1 mg·L−1,HRT为1 h为生物吸附池的运行条件。经过生物吸附池处理后,80%左右的COD被去除,有利于后续处理。
MBBR池具有较高的污泥浓度,池中填料上生长的微生物一直处于好氧环境,且没有泥龄的限制,填料上会不断富集专性好氧菌,有效提升了MBBR池对COD的去除效果。在装置运行期间调整了MBBR池的HRT和DO。HRT过短时生物降解就会不够充分,过长则会提高运行成本。DO过低会影响MBBR池的生物降解和硝化作用,DO过高可能会引起填料流化波动太大,使得一部分生物膜脱落,造成生物膜量减少,进而影响污染物的去除。由图2(b)与表2可知,在MBBR池的HRT从8 h增加至10 h时,MBBR池出水COD值下降约70 mg·L−1,去除率提高约23%。在相同HRT的情况下,DO浓度从3 mg·L−1增加至5 mg·L−1,MBBR池出水COD值约下降40 mg·L−1,去除率提高14%左右。
反应器运行58 d后,在纯硫自养反硝化滤池后,加装了活性焦柱,以进一步降低出水指标浓度。活性焦拥有较大的比表面积和丰富的中孔结构,能吸附污水中结构复杂的大分子物质,尤其是芳香族污染物[28]。活性焦柱的HRT分别从2 h降至1.5 h,HRT继续下降并未导致出水COD值提升,活性焦的处理效果较为稳定。经过活性焦吸附后,组合工艺出水COD稳定在30 mg·L−1以下,实现了稳定达标排放。
-
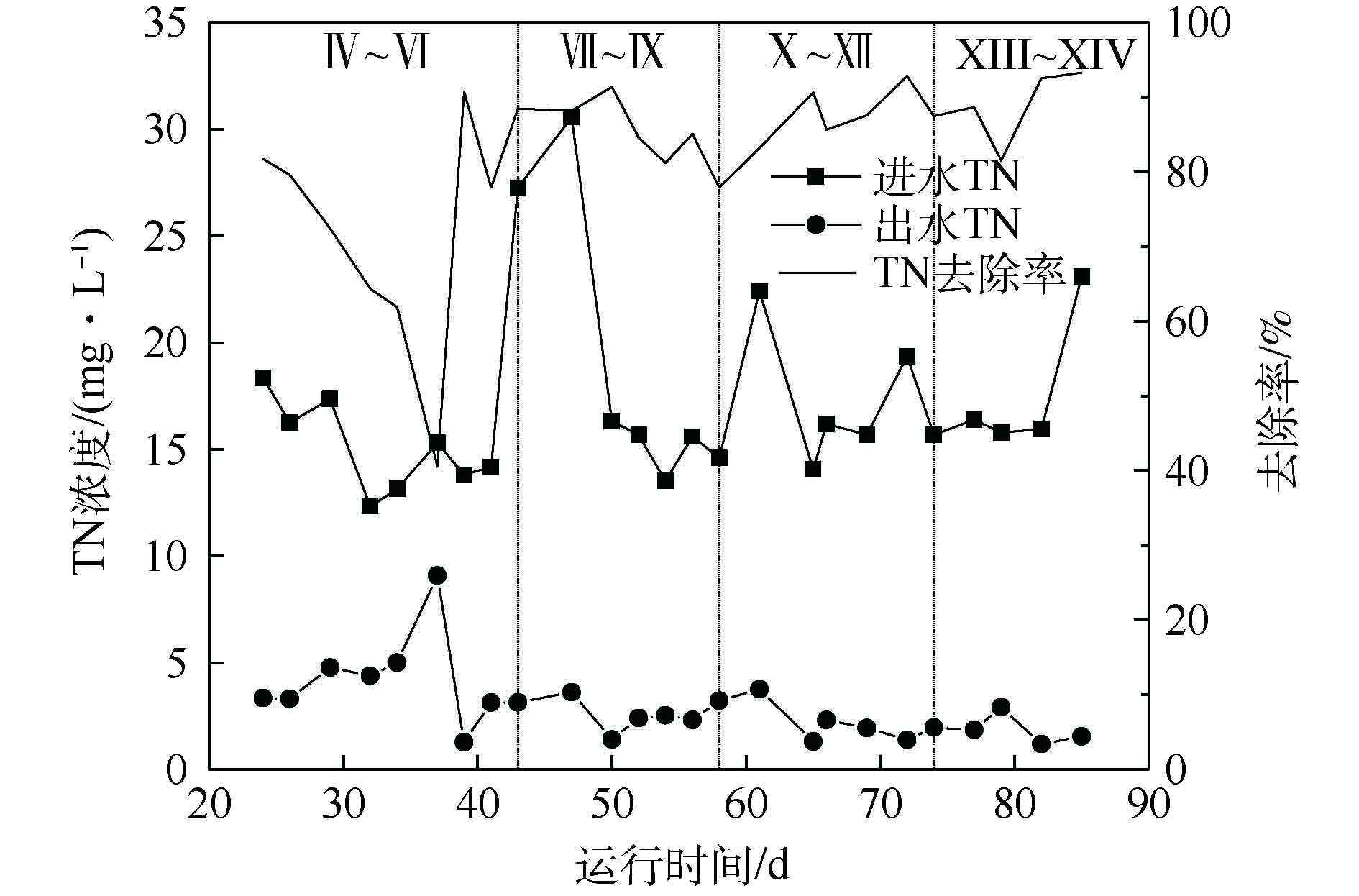
组合工艺是通过好氧MBBR池将污水中的氨氮转化为硝态氮,再由硫铁自养反硝化滤池将硝态氮去除,从而达到脱氮的目的。组合工艺对进水中氮的去除效果如图3所示。进水TN浓度范围为12~30 mg·L−1,平均值为16.92 mg·L−1。在装置进入优化阶段后,出水TN浓度稳定维持在5 mg·L−1以下。唯一的波动值出现在37 d,其原因是在35 d时将硫铁自养反硝化滤池改为纯硫自养反硝化滤池,装置处于调试稳定阶段,因此,出水TN出现波动,但出水仍可达到排放标准。
反应器各阶段
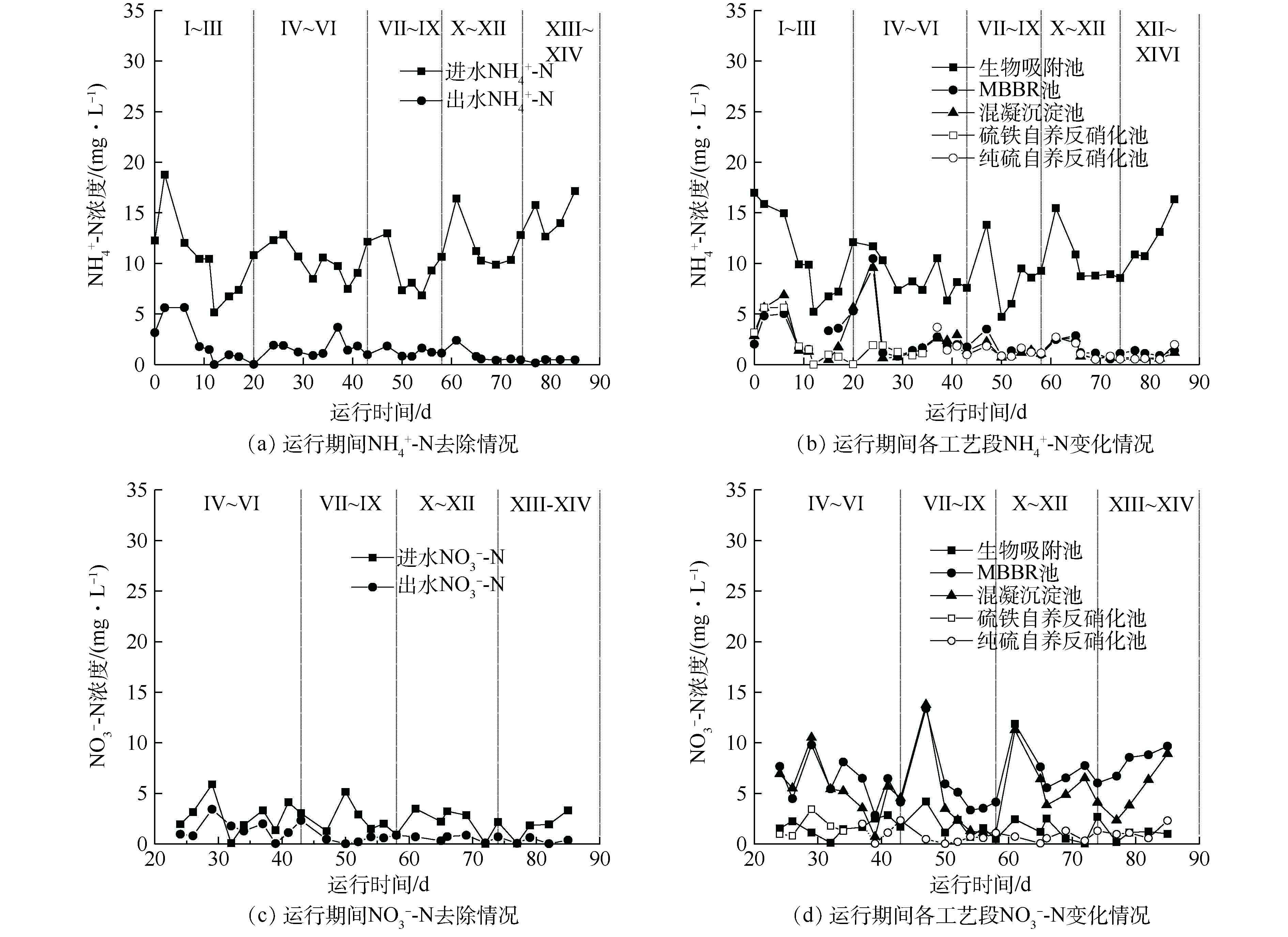
NH+4 -N和NO−3 -N的变化情况如图4所示。进水NH+4 -N和NO−3 -N浓度均值分别为10.25 mg·L−1和2.48 mg·L−1。在装置启动完成后,装置出水NH+4 -N浓度稳定在4 mg·L−1以下,NO−3 -N浓度基本维持在较低水平。由图4(b)和图4(d)可知,调整生物吸附池的参数对氨氮的去除影响较小,氮的转化主要在MBBR池和硫铁自养反硝化滤池。经过生物吸附池处理后,污水进入MBBR池进行硝化反应,将
NH+4 -N转化为NO−3 -N。经过第1阶段的优化后,MBBR池的HRT从8 h增加至10 h。HRT的增加给予了硝化菌充分的反应时间,MBBR池对NH+4 -N的去除效果明显提升,NH+4 -N浓度快速下降,其浓度从5 mg·L−1降至1 mg·L−1左右。继续增加MBBR池的HRT,NH+4 -N的去除效果没有明显的提升。硫铁自养反硝化滤池兼顾脱氮和除磷功能,脱氮硫杆菌以单质硫为电子供体将硝酸盐氮还原为氮气,铁屑析出的Fe3+与PO3−4 -P结合,实现水中磷的去除[29-30]。在前期运行过程中,发现硫铁自养反硝化滤池对磷的去除效果不稳定,因此,将硫铁自养反硝化滤池调整为纯硫自养反硝化滤池。纯硫自养反硝化滤池与硫铁自养反硝化滤池相比,对NO−3 -N的去除效果稳定和高效,但没有除磷功能。当纯硫自养反硝化滤池的HRT从4 h减少3 h时,出水NO−3 -N浓度没有明显提升,但当HRT从3 h继续降低至2 h时,出水NO−3 -N浓度从0.5 mg·L−1上升至2 mg·L−1。其原因可能是较快的流速使反硝化菌没有足够的反应时间,导致NO−3 -N的去除效果不佳。较快的水流冲刷滤池,还会使得较松散和老化的生物膜不断脱落和被冲走,造成部分微生物解体死亡并且释放自身体内的NH+4 -N。故选择3 h作为纯硫自养反硝化滤池的运行参数。在NH+4 -N和NO−3 -N均被有效去除的情况下,TN出水浓度在稳定状态下始终在5 mg·L−1以下。 -
组合工艺进水TP在4~12 mg·L−1之间波动,其中,
PO3−4 -P/TP的比值较高,除磷药剂对PO3−4 -P有较好地去除效果,因此,组合工艺采用在混凝沉淀池中投加除磷药剂结合硫铁自养反硝化滤池,可实现对磷的有效去除。如图5(a)所示,在0~50 d的运行期间内,出水PO3−4 -P浓度在1 mg·L−1左右波动,且波动较大。其原因是:组合工艺通过混凝沉淀池投加25 mg·L−1的PAC与硫铁自养反硝化联合除磷,但25 mg·L−1的PAC投加量未能满足PO3−4 -P的高效去除。如图5(b)所示,在经过混凝沉淀池后,硫铁自养反硝化滤池的出水PO3−4 -P出现高于混凝沉淀池的现象,推测原因为硫铁自养反硝化滤池出水中携带从生物膜中脱落下的微生物分解产物。故可将硫铁反硝化滤池调整为纯硫反硝化滤池,以除磷药剂作为PO3−4 -P的主要去除手段。为筛选除磷药剂的种类与投加量,进行了混凝沉淀池的除磷药剂的比选实验,分别采用聚合氯化铝(PAC)、聚合氯化铝铁(PAFC)和双酸铝铁(PAFCS)。由图6可知,3种除磷药剂的效果为:PAFCS>PAC>PAFC。其中,PAFCS对
PO3−4 -P的去除效果明显,其在0.5‰的投加量的情况下,出水PO3−4 -P的浓度降至0.1 mg·L−1,PAC的投加量在100 mg·L−1时能达到相同的效果。在达到相同处理效果的条件下,PAFCS的成本远高于PAC。因此,选择PAC作为最佳的除磷药剂,为保证出水TP稳定达标,采用100 mg·L−1作为投加量。在运行50 d后,采用投加100 mg·L−1的PAC作为除磷手段。由图5(a)和图5(b)可知,出水TP浓度下降明显。在优化阶段3时,系统加入了活性焦柱,出水TP进一步下降,出水稳定达标排放。 -
在58 d稳定运行后,在纯硫自养反硝化滤池后端连接活性焦柱,以提高组合工艺对COD及色度的去除效果。组合工艺运行效果如图7所示。印染废水的进水色度在250倍左右,经过生物吸附池和MBBR池处理后,废水色度显著降低,出水色度降至60倍左右。活性焦柱将色度基本完全吸附,组合工艺出水色度在2倍以下。印染废水中的色度主要是由显色基团(—N=N、—N=O等)及助色基团(—OH、—NH2、—COOH等)构成,生物吸附池与MBBR池均对色度有去除效果,生物吸附通过快速沉降进水中大量的污染物从而去除色度。在MBBR池中填料和泥水混合液在充氧流化状态下与废水充分反应,大大增加了微生物与污染物质的接触时间,有利于对发色基团进行氧化,提高了脱色效率[31]。活性焦具有丰富的孔结构,同时还具有丰富的有机官能团,如羟基、酚羟基、混型羟基等,能够通过物理吸附和化学吸附去除大量的发色基团,因此,组合工艺出水色度基本完全去除。
-
在运行了85 d后,最终稳定出水如表3所示。各项指标均符合《太湖地区城镇污水处理厂及重点工业行业主要污染物排放限制》(DB 32/1072-2018)中对纺织染整行业的排放标准。
-
1)生物吸附池主要进行有机物的快速吸附与沉降,在DO为1 mg·L−1,HRT为1 h时,COD的去除率达到80%以上。
2)MBBR池是利用活性污泥去除可生物降解的COD和将
NH+4 -N转化为硝态氮,在DO浓度为5 mg·L−1和HRT在10 h时,COD与NH+4 -N的去除率均达到90%以上;当PAC的投加量达到100 mg·L−1时,混凝沉淀池对PO3−4 -P的去除率达到85%以上。3)纯硫自养反硝化滤池对
NO−3 -N的去除效果优于硫铁自养反硝化滤池,在HRT为3 h时,出水NO−3 -N浓度稳定在0.5 mg·L−1以下,出水NO−3 -N基本完全去除。4)活性焦柱利用其较大的比表面积与丰富的中孔结构,可有效吸附进水中含有的SS、色度和有机物等污染物,经过活性焦处理后,组合工艺出水指标浓度进一步下降,出水COD、
NH+4 -N、TP和TN浓度分别为16、0.56、0.32和1.39 mg·L−1,实现了印染废水出水的稳定高标准排放。
印染废水高标准排放组合工艺优化
High standard discharge combination process optimization for printing and dyeing wastewater treatment
-
摘要: 为了实现印染废水的高标准排放,构建了生物吸附/MBBR/混凝沉淀池/硫铁自养反硝化/活性焦组合工艺,并对其进行了优化运行研究;考察了不同水力停留时间(HRT)和溶解氧(DO)对系统污染物去除的影响。结果表明:生物吸附池和MBBR池的HRT分别为1 h和10 h、DO分别为1 mg·L−1和5 mg·L−1的情况下,污染物的去除效果最佳;其中,COD的去除率达到98%;在最优条件下,组合工艺出水COD、
NH+4 -N、TP和TN浓度分别为 16、0.56、0.32和1.39 mg·L−1,污水色度基本完全去除。该组合工艺实现了印染废水的高标准排放,为印染废水处理的工程应用提供了数据和技术支撑。Abstract: In order to achieve high standard discharge of printing and dyeing wastewater, a biosorption/MBBR/coagulation sedimentation tank/sulfur iron autotrophic denitrification/active coke combination process was constructed, and its operation was also optimized. The effects of hydraulic retention time (HRT) and dissolved oxygen (DO) on pollutant removal by this combination system were investigated. The results showed that the best pollutant removal effect was achieved at HRTs of 1 h and 10 h, DO values of 1 mg·L−1 and 5 mg·L−1 for the biosorption and the MBBR tanks, respectively, the COD removal rate could reach 98%. Under the optimal conditions, the COD, NH4+-N, TP and TN concentrations of the combined process effluent were 16, 0.56, 0.32 and 1.39 mg·L−1, respectively, and the color of the wastewater was almost completely removed. The combined process achieves high standard discharge of printing and dyeing wastewater, and provides data and technical support for engineering applications of printing and dyeing wastewater treatment. -

图 1 生物吸附/MBBR/混凝沉淀/硫铁自养反硝化/活性焦组合工艺
Figure 1. Combination process of biosorption/MBBR/coagulation sedimentation/sulfur iron autotrophic denitrification/active coke
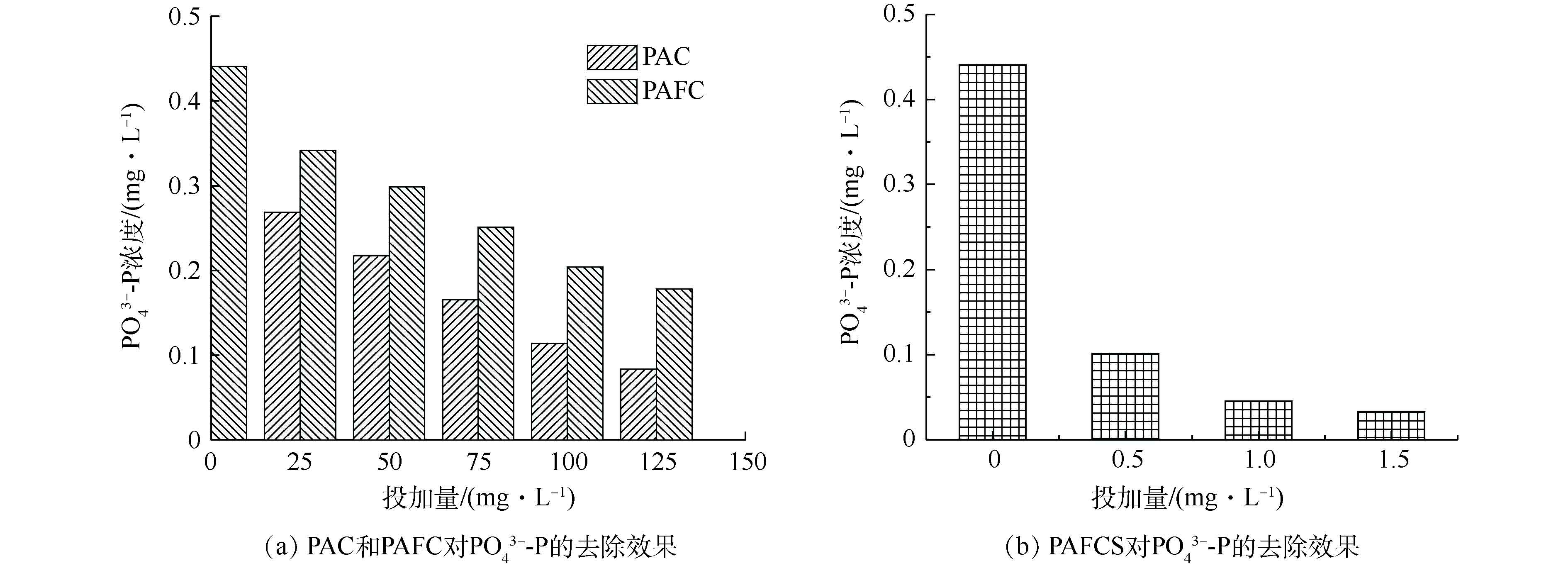
图 6 混凝沉淀池除磷药剂的比选
Figure 6. Comparison of phosphorus removal agent in coagulation sedimentation tank
表 1 进水主要水质指标范围
Table 1. Influent water quality index range
COD/(mg·L−1) BOD/(mg·L−1) TP/(mg·L−1) TN/(mg·L−1) SS/(mg·L−1) pH 色度/倍 300~800 100~300 3.38 19.36 300~800 8.0-8.5 256  下载: 导出CSV
下载: 导出CSV
表 2 组合工艺运行条件
Table 2. Operation conditions of the combined process
运行阶段 阶段序号 运行时间/d 运行参数 生物吸附池 MBBR池 硫铁自养反硝化池 活性焦柱HRT/h DO/(mg·L−1) HRT/h DO/(mg·L−1) HRT/h 硫铁体积比 HRT/h 启动阶段 Ⅰ 0~6 0.5 0.5 3 8 5∶1 4 — Ⅱ 7~17 0.5 0.5 3 8 5∶1 15 — Ⅲ 18~20 0.5 0.5 3 8 5∶1 4 — 优化阶段1 Ⅳ 21~29 0.5 1 3 8 5∶1 4 — Ⅴ 30~37 0.5 1 3 10 5∶1 4 — Ⅵ 38~43 0.5 1.5 3 10 5∶1 4 — 优化阶段2 Ⅶ 44~50 1 1 5 8 — 4 — Ⅷ 51~54 1 1 5 10 — 4 — Ⅸ 55~58 1 1.5 5 10 — 4 — Ⅹ 58~65 1 1 5 12 — 4 2 优化阶段3 Ⅺ 66~68 1 1 5 10 — 4 2 Ⅻ 69~73 2 1 5 10 — 4 2 XIII 74~79 1 1 7 10 — 4 2 优化阶段4 XIV 80~82 1 1 5 10 — 3 1.5 XV 83~85 1 1 5 10 — 2 1 注:在50 d后,将硫铁自养反硝化柱调整为纯硫自养反硝化柱,因此,无硫铁体积比的数据,活性焦柱于第61天开始运行。
下载: 导出CSV
表 3 组合工艺出水水质与排放标准
Table 3. Effluent water quality of the combined process and discharge standards
水质数据来源 COD/(mg·L−1) TN/(mg·L−1) NH+4 -N/(mg·L−1)</td> <td class="table_top_border" align="center" valign="middle">TN/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{NH}}_4^{+} $<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead> <tr> <td class="table_top_border" align="center" valign="middle">水质数据来源</td> <td class="table_top_border" align="center" valign="middle">COD/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle">TN/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{NH}}_4^{+} $<alternatives><img class="graphic" src="201811149qiankai_Z-20190628102625.jpg"><img class="graphic" src="201811149qiankai_Z-20190628102625.png"></alternatives></inline-formula>-N/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle">TP/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle">色度/倍</td> </tr> </thead>
<tbody> <tr> <td class="table_top_border2" align="center" valign="middle">本研究中组合工艺出水</td> <td class="table_top_border2" align="center" valign="middle">16</td> <td class="table_top_border2" align="center" valign="middle">1.39</td> <td class="table_top_border2" align="center" valign="middle">0.56</td> <td class="table_top_border2" align="center" valign="middle">0.32</td> <td class="table_top_border2" align="center" valign="middle">2</td> </tr> <tr> <td class="table_bottom_border" align="center" valign="middle">出水排放标准</td> <td class="table_bottom_border" align="center" valign="middle">60</td> <td class="table_bottom_border" align="center" valign="middle">12</td> <td class="table_bottom_border" align="center" valign="middle">5</td> <td class="table_bottom_border" align="center" valign="middle">0.5</td> <td class="table_bottom_border" align="center" valign="middle">30</td> </tr> </tbody>
</table></div></foreignObject></svg>"></inline-formula>-N/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle">TP/(mg·L<sup>−1</sup>)</td> <td class="table_top_border" align="center" valign="middle">色度/倍</td> </tr> </thead>
<tbody> <tr> <td class="table_top_border2" align="center" valign="middle">本研究中组合工艺出水</td> <td class="table_top_border2" align="center" valign="middle">16</td> <td class="table_top_border2" align="center" valign="middle">1.39</td> <td class="table_top_border2" align="center" valign="middle">0.56</td> <td class="table_top_border2" align="center" valign="middle">0.32</td> <td class="table_top_border2" align="center" valign="middle">2</td> </tr> <tr> <td class="table_bottom_border" align="center" valign="middle">出水排放标准</td> <td class="table_bottom_border" align="center" valign="middle">60</td> <td class="table_bottom_border" align="center" valign="middle">12</td> <td class="table_bottom_border" align="center" valign="middle">5</td> <td class="table_bottom_border" align="center" valign="middle">0.5</td> <td class="table_bottom_border" align="center" valign="middle">30</td> </tr> </tbody>
</table></div></foreignObject></svg>)
TP/(mg·L−1) 色度/倍 本研究中组合工艺出水 16 1.39 0.56 0.32 2 出水排放标准 60 12 5 0.5 30
下载: 导出CSV
-
[1] 周贤波, 李晓. 印染废水物化处理技术研究[J]. 应用化工, 2018, 47(5): 1058-1061. doi: 10.3969/j.issn.1671-3206.2018.05.051 [2] 范剑. 苏浙两省污水排放标准差异对浙江印染产业发展影响的分析[J]. 统计科学与实践, 2012(7): 16-17. doi: 10.3969/j.issn.1674-8905.2012.07.005 [3] 奚旦立, 陈季华. 在保护环境的前提下发展印染业由太湖蓝藻水污染事件引发的思考[J]. 纺织服装周刊, 2007(41): 19. [4] 尤近仁. 印染产业的转型升级与集聚发展[J]. 纺织导报, 2015(4): 22-24. doi: 10.3969/j.issn.1003-3025.2015.04.006 [5] 童坤, 孙伟, 陈雯. 长江经济带水环境保护及治理政策比较研究[J]. 区域与全球发展, 2019, 3(1): 5-16. [6] 张勇, 侯得印, 赵长伟, 等. 超滤-膜接触臭氧氧化技术处理印染废水[J]. 环境工程学报, 2017, 11(6): 3363-3368. [7] 王东, 王小东, 朱引, 等. 生物吸附-多级A/O-活性焦组合工艺对污水中氮磷及有机污染物的去除[J]. 环境工程学报, 2018, 12(7): 1907-1916. [8] 钟华文, 林培喜, 谢文玉, 等. 混凝与AB法联用处理制革废水[J]. 工业用水与废水, 2011, 42(2): 30-32. doi: 10.3969/j.issn.1009-2455.2011.02.008 [9] NIU K S, JIANG Y M, WEI W, et al. Problems and countermeasures of AB process in Zibo Wastewater Treatment Plant[J]. Water & Wastewater Engineering, 2004, 30(2): 27-29. [10] 陈金灿. MBBR-超滤-催化氧化组合工艺处理工业废水的中试研究[J]. 广东化工, 2018, 45(6): 170-172. doi: 10.3969/j.issn.1007-1865.2018.06.074 [11] CASTRO F D, JOÁO PAULO B, MÁRCIA D. Treatment of a simulated textile wastewater containing the reactive orange 16 azo dye by a combination of ozonation and moving-bed biofilm reactor: Evaluating the performance, toxicity, and oxidation by-products[J]. Environmental Science & Pollution Research, 2017, 24(7): 1-10. [12] YANG X, MARTI C, VICTORR L G. A review on the present situation of wastewater treatment in textile industry with membrane bioreactor and moving bed biofilm reactor[J]. Desalination & Water Treatment, 2018, 103: 315-322. [13] 王广智, 王敬元, 黄丽坤, 等. 好氧MBBR处理含吲哚废水的工艺优化试验研究[J]. 环境保护科学, 2017, 43(2): 41-46. [14] 徐忠强, 郝瑞霞, 徐鹏程, 等. 硫铁填料和微电流强化再生水脱氮除磷的研究[J]. 中国环境科学, 2016, 36(2): 406-413. doi: 10.3969/j.issn.1000-6923.2016.02.016 [15] SAHINKAYA E, KILIC A. Heterotrophic and elemental-sulfur-based autotrophic denitrification processes for simultaneous nitrate and Cr(VI) reduction[J]. Water Research, 2014, 50: 278-286. doi: 10.1016/j.watres.2013.12.005 [16] 周彦卿, 郝瑞霞, 刘思远, 等. 新型硫铁复合填料强化再生水深度脱氮除磷[J]. 环境科学, 2017, 38(10): 4309-4315. [17] LV X, SHAO M, LI J, et al. Nitrate removal with lateral flow sulphur autotrophic denitrification reactor[J]. Environmental Technology Letters, 2014, 35(21): 2692-2697. doi: 10.1080/09593330.2014.918660 [18] 彭军. 混凝沉淀/活性污泥法处理印染工业园废水[J]. 广东化工, 2018, 45(18): 139-140. doi: 10.3969/j.issn.1007-1865.2018.18.066 [19] 张萍, 胡志祥, 冯军, 等. BOC/深水氧化沟/混凝沉淀技术处理印染废水[J]. 染整技术, 2017, 39(11): 39-42. doi: 10.3969/j.issn.1005-9350.2017.11.010 [20] 胡溪, 禹晓磊, 崔粲粲, 等. 活性焦技术在印染废水处理中的实验研究[J]. 水处理技术, 2014, 40(2): 75-78. [21] 李欣珏. 活性炭吸附对印染废水生化出水中各类有机物去除特性研究[D]. 上海: 华东理工大学, 2012. [22] 国家环境保护局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [23] 李兴. 印染废水生物处理系统活性污泥性质的检测方法比较研究[D]. 西安: 西安工程大学, 2011. [24] 孙凯. 应对进水异常的厌氧/缺氧/好氧污水处理改造技术研究[D]. 北京: 清华大学, 2015. [25] 张芝利. 生物吸附法对水中重金属离子的处理研究[D]. 西安: 西安工程大学, 2011. [26] 王雄. 生物吸附法预处理腈纶废水及氨氮降解试验研究[D]. 兰州: 兰州交通大学, 2015. [27] 刘绍根. 城市污水生物絮凝吸附工艺的特性及模拟研究[D]. 合肥: 中国科学技术大学, 2010. [28] 李全. 流化床制活性焦用于水处理的研究[J]. 煤炭转化, 2008(2): 74-77. doi: 10.3969/j.issn.1004-4248.2008.02.018 [29] KONG Z, LI L, FENG C, et al. Comparative investigation on integrated vertical-flow biofilters applying sulfur-based and pyrite-based autotrophic denitrification for domestic wastewater treatment[J]. Bioresource Technology, 2016, 211: 125-135. doi: 10.1016/j.biortech.2016.03.083 [30] 袁玉玲, 李睿华. 硫磺/石灰石自养反硝化系统脱氮除磷性能研究[J]. 环境科学, 2011, 32(7): 2041-2046. [31] 牟淑杰. PDMDAAC-MBBR组合工艺处理印染废水试验研究[J]. 印染助剂, 2009, 26(11): 39-41. doi: 10.3969/j.issn.1004-0439.2009.11.009 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4670
- HTML全文浏览数: 4670
- PDF下载数: 65
- 施引文献: 0
